La fiebre es un motivo de consulta muy frecuente, especialmente en los
niños. Durante mucho tiempo, y todavía persiste esta idea, hemos
aconsejado el uso de paracetamol e ibuprofeno de forma alternante en el
tratamiento de la fiebre en los niños (y en los adultos). Cambiar
primero en nuestra cabeza y luego en la de los preocupados padres esta
práctica es difícil; a menudo te preguntan sobre este aspecto. Sin
embargo, desde hace tiempo no se aconseja proceder de esta manera; os
dejo un artículo
en castellano que ahonda en esta materia. Y ya puestos con la fiebre en
los niños, también merecen la pena estas dos hojas informativas de la i-botika sobre los dos antitérmicos referidos en el uso pediátrico: paracetamol e ibuprofeno,
creo que son una joyita y hace tiempo que las tengo pegadas en el
corcho de mi consulta, seguro que más de uno de vosotros también lo ha
hecho.
Os recordamos que hace ya mucho, muchísimo, publicamos esta entrada sobre el tratamiento de la fiebre que tal vez os apetezca releer.
martes, 31 de marzo de 2015
Docencia en Algemesí, Incontinencia Urinaria.
A continuación os dejo el resumen del documento de consenso elaborado en 2013 por las principales sociedades de atención primaria nacionales y por la Asociación Española de Urología para el abordaje de la incontinencia urinaria, un problema de salud que afecta a gran cantidad de personas en España, y para el que las medidas no farmacológicas pueden ayudar a resolver/mejorar una gran parte de los casos.
lunes, 30 de marzo de 2015
Salud y fármacos. Síndrome de piernas inquietas: sobrediagnosticado y sobretratado.
(Restless Legs Syndrome: Overdiagnosed And Overtreated)
Wort Pills, Best Pills 2014: 20 (12)
Traducido por Salud y Fármacos
Cada cierto tiempo, es posible que la mayoría de la gente se sienta inquieta por la noche y le cueste quedarse dormido. Pero si su inquietud se debe específicamente a una urgencia incontrolable de mover uno o más miembros, es posible que le diagnostiquen un trastorno conocido como síndrome de piernas inquietas (SPI).
El SPI no se ha reconocido formalmente como un trastorno médico hasta hace poco tiempo, y dicha designación ha sido polémica. El diagnóstico se basa únicamente en síntomas vagos y subjetivos que a menudo también se observan en algunos trastornos psiquiátricos y físicos, y su causa en la mayoría de los pacientes sigue siendo desconocida.
Asimismo, la introducción del SPI como enfermedad coincidió con una intensa campaña promocional por parte de la industria farmacéutica para explotar la incertidumbre y la falta de conocimiento sobre este trastorno y expandir el mercado de fármacos lucrativos de la industria. Por este motivo nos preguntamos si la mayoría de los pacientes con SPI, especialmente aquellos con síntomas leves, están realmente afectados por dicho trastorno.
Esto es particularmente relevante al considerar las opciones terapéuticas. Los medicamentos aprobados por la FDA para el tratamiento del SPI han demostrado ser algo efectivos únicamente a corto plazo y presentan varios efectos secundarios graves, incluyendo, en muchos casos, el agravamiento de los síntomas que pretenden tratar.
Algunas evidencias sugieren que el trastorno nace de una disfunción en el área del cerebro responsable de la coordinación de los movimientos musculares. La deficiencia de hierro y, específicamente, los niveles bajos de hierro en ciertas áreas del cerebro también se han asociado con el SPI.
La Fundación Nacional del Sueño clasifica el SPI en dos tipos. El SPI primario se diagnostica en aquellos sujetos con una sospecha de causa genética o en aquellos en los que se desconoce la causa, mientras que el SPI secundario se diagnostica cuando se identifica una causa potencialmente reversible. Los pacientes que muestran síntomas de SPI antes de los 40 años tienen más probabilidades de tener antecedentes familiares de la enfermedad, y por tanto un posible componente genético.
El SPI secundario se ha asociado con trastornos como enfermedad renal crónica, diabetes y neuropatía periférica. Las mujeres en el último trimestre de embarazo también pueden presentar un riesgo mayor de SPI secundario. Se relaciona ciertos medicamentos con la presencia o agravamiento del SPI secundario. Entre estos se incluyen fármacos antinauseosos, antipsicóticos, antidepresivos y antihistamínicos para el resfriado o la alergia.
Es probable que factores psicológicos y del estilo de vida jueguen un papel importante en el desarrollo o en la gravedad de todas las formas del SPI. El consumo de alcohol y cafeína, la falta de sueño o un horario de sueño irregular, y trastornos del ánimo como depresión y ansiedad se han asociado con los síntomas del SPI.
Manejo terapéutico del SPI
Generalmente en los pacientes diagnosticados con el SPI se evalúan las causas secundarias. Debe efectuarse un escrutinio pormenorizado de los medicamentos que consume el paciente para determinar si los síntomas son inducidos por los fármacos. Si no se halla una causa subyacente de los síntomas, probablemente el paciente presente SPI primario.
Solo debe procederse a la terapia farmacológica cuando los síntomas del SPI persistan a pesar de los cambios del estilo de vida, y en casos graves e incapacitantes. Actualmente existen fármacos aprobados para el tratamiento del SPI: los agonistas dopaminérgicos pramipexol (Mirapex), ropinirol (Requip) y el parche transdérmico rotigotina (Neupro), así como gabapentina enacarbil de liberación prolongada (Horizant). Todos estos fármacos se aprobaron únicamente para casos moderados o graves de SPI primario.
No obstante, algunos médicos prescriben otros fármacos no aprobados por la FDA para el tratamiento del SPI fuera de las indicaciones autorizadas. Estos medicamentos, como antiepilépticos, sedantes benzodiacepínicos y fármacos dopaminérgicos, presentan efectos secundarios graves y en ocasiones mortales y la FDA nunca ha evaluado favorablemente su efectividad para el tratamiento del SPI.
Limitaciones de las terapias farmacológicas
Por desgracia, todos los medicamentos aprobados actualmente para el SPI solo han demostrado su eficacia a corto plazo. Una revisión de 2012 de todos los ensayos clínicos aleatorizados de agonistas dopaminérgicos para el tratamiento del SPI, incluyendo aquellos no aprobados por la FDA para este trastorno, halló que en los ensayos los sujetos recibieron tratamiento durante solo 10 semanas y ningún ensayo superó los siete meses de duración. El único agonista no dopaminérgico aprobado para el tratamiento del SPI, gabapentina enacarbil de liberación prolongada, se aprobó a partir de dos ensayos de 12 semanas de duración.
Una de las complicaciones principales a largo plazo de los agonistas dopaminérgicos en el SPI es su potencial para generar dependencia física en los pacientes, lo cual requerirá que con el trascurso del tiempo se tenga que ir aumentando la dosis. Se piensa que este fenómeno, conocido como augmentación, afecta a un 7% de los pacientes tratados por cada año de tratamiento. Generalmente los pacientes que desarrollan augmentación por agonistas dopaminérgicos interrumpen su tratamiento, pero muchos experimentan posteriormente un agravamiento de los síntomas del SPI en comparación con el inicio del tratamiento. Este hecho plantea la pregunta de si cuando se emplean de forma crónica los agonistas dopaminérgicos hacen más mal que bien.
Otros efectos secundarios de los tres agonistas dopaminérgicos aprobados para el SPI incluyen hipotensión a niveles peligrosos en bipedestación, alucinaciones y psicosis. Un estudio reciente que analizó cientos de informes de eventos adversos enviados a la FDA concluyó que el juego patológico, la hipersexualidad y las compras compulsivas son efectos secundarios probables del uso de agonistas dopaminérgicos en algunos pacientes con SPI y enfermedad de Parkinson. En 2012, la FDA advirtió que pramipexol puede producir un ligero aumento del riesgo de insuficiencia cardiaca congestiva. Gabapentina enacarbil de liberación prolongada puede producir pensamientos suicidas. Los cuatro fármacos pueden producir somnolencia diurna excesiva y repentina, lo que puede afectar peligrosamente a la capacidad de conducción.
Lo que se puede hacer
Si presenta síntomas del SPI, como molestias o inquietud en las piernas por la noche, consulte a su médico sobre si presenta un trastorno o si está tomando un medicamento que pudiera causar dichos síntomas.
Si sus síntomas no son incapacitantes, antes de recibir tratamiento intente los cambios en su dieta y en su rutina diaria que se recomiendan en este artículo.
Si usted o su médico deciden que es necesario el tratamiento farmacológico para este trastorno, asegúrese de revisar los efectos secundarios de cada tratamiento y tenga en cuenta que todos los fármacos aprobados por la FDA para tratar el SPI han demostrado la mejora de los síntomas solo a corto plazo. Nunca trate este trastorno con un fármaco que la FDA no ha aprobado para tal fin.
Wort Pills, Best Pills 2014: 20 (12)
Traducido por Salud y Fármacos
Cada cierto tiempo, es posible que la mayoría de la gente se sienta inquieta por la noche y le cueste quedarse dormido. Pero si su inquietud se debe específicamente a una urgencia incontrolable de mover uno o más miembros, es posible que le diagnostiquen un trastorno conocido como síndrome de piernas inquietas (SPI).
El SPI no se ha reconocido formalmente como un trastorno médico hasta hace poco tiempo, y dicha designación ha sido polémica. El diagnóstico se basa únicamente en síntomas vagos y subjetivos que a menudo también se observan en algunos trastornos psiquiátricos y físicos, y su causa en la mayoría de los pacientes sigue siendo desconocida.
Asimismo, la introducción del SPI como enfermedad coincidió con una intensa campaña promocional por parte de la industria farmacéutica para explotar la incertidumbre y la falta de conocimiento sobre este trastorno y expandir el mercado de fármacos lucrativos de la industria. Por este motivo nos preguntamos si la mayoría de los pacientes con SPI, especialmente aquellos con síntomas leves, están realmente afectados por dicho trastorno.
Esto es particularmente relevante al considerar las opciones terapéuticas. Los medicamentos aprobados por la FDA para el tratamiento del SPI han demostrado ser algo efectivos únicamente a corto plazo y presentan varios efectos secundarios graves, incluyendo, en muchos casos, el agravamiento de los síntomas que pretenden tratar.
Sobre el SPI
Según los National Institutes of Health (NIH) de EE UU, el SPI “es un trastorno neurológico caracterizado por movimientos rítmicos y/o insidiosos, tirones u otras sensaciones molestas en las piernas y una urgencia incontrolable, y en ocasiones incontenible, de moverlas”. Los síntomas del SPI son:
Según los National Institutes of Health (NIH) de EE UU, el SPI “es un trastorno neurológico caracterizado por movimientos rítmicos y/o insidiosos, tirones u otras sensaciones molestas en las piernas y una urgencia incontrolable, y en ocasiones incontenible, de moverlas”. Los síntomas del SPI son:
- Empeora por la noche y mejora por la mañana.
- Se desencadena en el descanso, la relajación o el sueño.
- Mejora, y se mantiene mejor, con el movimiento.
Algunas evidencias sugieren que el trastorno nace de una disfunción en el área del cerebro responsable de la coordinación de los movimientos musculares. La deficiencia de hierro y, específicamente, los niveles bajos de hierro en ciertas áreas del cerebro también se han asociado con el SPI.
La Fundación Nacional del Sueño clasifica el SPI en dos tipos. El SPI primario se diagnostica en aquellos sujetos con una sospecha de causa genética o en aquellos en los que se desconoce la causa, mientras que el SPI secundario se diagnostica cuando se identifica una causa potencialmente reversible. Los pacientes que muestran síntomas de SPI antes de los 40 años tienen más probabilidades de tener antecedentes familiares de la enfermedad, y por tanto un posible componente genético.
El SPI secundario se ha asociado con trastornos como enfermedad renal crónica, diabetes y neuropatía periférica. Las mujeres en el último trimestre de embarazo también pueden presentar un riesgo mayor de SPI secundario. Se relaciona ciertos medicamentos con la presencia o agravamiento del SPI secundario. Entre estos se incluyen fármacos antinauseosos, antipsicóticos, antidepresivos y antihistamínicos para el resfriado o la alergia.
Es probable que factores psicológicos y del estilo de vida jueguen un papel importante en el desarrollo o en la gravedad de todas las formas del SPI. El consumo de alcohol y cafeína, la falta de sueño o un horario de sueño irregular, y trastornos del ánimo como depresión y ansiedad se han asociado con los síntomas del SPI.
Manejo terapéutico del SPI
Generalmente en los pacientes diagnosticados con el SPI se evalúan las causas secundarias. Debe efectuarse un escrutinio pormenorizado de los medicamentos que consume el paciente para determinar si los síntomas son inducidos por los fármacos. Si no se halla una causa subyacente de los síntomas, probablemente el paciente presente SPI primario.
A menudo los síntomas de los
pacientes con SPI leve o moderada pueden reducirse o resolverse con
cambios en su estilo de vida o en su rutina diaria. Las estrategias
recomendadas por el NIH para reducir los síntomas del SPI incluyen:
- Reducción del consumo de cafeína, alcohol o tabaco.
- Mejorar la higiene del sueño, incluyendo mantener un patrón regular de sueño.
- Realizar ejercicio con regularidad.
- Aplicar tratamientos a las piernas, como masajes, baños calientes o emplear bolsas de calor o frío.
Solo debe procederse a la terapia farmacológica cuando los síntomas del SPI persistan a pesar de los cambios del estilo de vida, y en casos graves e incapacitantes. Actualmente existen fármacos aprobados para el tratamiento del SPI: los agonistas dopaminérgicos pramipexol (Mirapex), ropinirol (Requip) y el parche transdérmico rotigotina (Neupro), así como gabapentina enacarbil de liberación prolongada (Horizant). Todos estos fármacos se aprobaron únicamente para casos moderados o graves de SPI primario.
No obstante, algunos médicos prescriben otros fármacos no aprobados por la FDA para el tratamiento del SPI fuera de las indicaciones autorizadas. Estos medicamentos, como antiepilépticos, sedantes benzodiacepínicos y fármacos dopaminérgicos, presentan efectos secundarios graves y en ocasiones mortales y la FDA nunca ha evaluado favorablemente su efectividad para el tratamiento del SPI.
Limitaciones de las terapias farmacológicas
Por desgracia, todos los medicamentos aprobados actualmente para el SPI solo han demostrado su eficacia a corto plazo. Una revisión de 2012 de todos los ensayos clínicos aleatorizados de agonistas dopaminérgicos para el tratamiento del SPI, incluyendo aquellos no aprobados por la FDA para este trastorno, halló que en los ensayos los sujetos recibieron tratamiento durante solo 10 semanas y ningún ensayo superó los siete meses de duración. El único agonista no dopaminérgico aprobado para el tratamiento del SPI, gabapentina enacarbil de liberación prolongada, se aprobó a partir de dos ensayos de 12 semanas de duración.
Una de las complicaciones principales a largo plazo de los agonistas dopaminérgicos en el SPI es su potencial para generar dependencia física en los pacientes, lo cual requerirá que con el trascurso del tiempo se tenga que ir aumentando la dosis. Se piensa que este fenómeno, conocido como augmentación, afecta a un 7% de los pacientes tratados por cada año de tratamiento. Generalmente los pacientes que desarrollan augmentación por agonistas dopaminérgicos interrumpen su tratamiento, pero muchos experimentan posteriormente un agravamiento de los síntomas del SPI en comparación con el inicio del tratamiento. Este hecho plantea la pregunta de si cuando se emplean de forma crónica los agonistas dopaminérgicos hacen más mal que bien.
Otros efectos secundarios de los tres agonistas dopaminérgicos aprobados para el SPI incluyen hipotensión a niveles peligrosos en bipedestación, alucinaciones y psicosis. Un estudio reciente que analizó cientos de informes de eventos adversos enviados a la FDA concluyó que el juego patológico, la hipersexualidad y las compras compulsivas son efectos secundarios probables del uso de agonistas dopaminérgicos en algunos pacientes con SPI y enfermedad de Parkinson. En 2012, la FDA advirtió que pramipexol puede producir un ligero aumento del riesgo de insuficiencia cardiaca congestiva. Gabapentina enacarbil de liberación prolongada puede producir pensamientos suicidas. Los cuatro fármacos pueden producir somnolencia diurna excesiva y repentina, lo que puede afectar peligrosamente a la capacidad de conducción.
Lo que se puede hacer
Si presenta síntomas del SPI, como molestias o inquietud en las piernas por la noche, consulte a su médico sobre si presenta un trastorno o si está tomando un medicamento que pudiera causar dichos síntomas.
Si sus síntomas no son incapacitantes, antes de recibir tratamiento intente los cambios en su dieta y en su rutina diaria que se recomiendan en este artículo.
Si usted o su médico deciden que es necesario el tratamiento farmacológico para este trastorno, asegúrese de revisar los efectos secundarios de cada tratamiento y tenga en cuenta que todos los fármacos aprobados por la FDA para tratar el SPI han demostrado la mejora de los síntomas solo a corto plazo. Nunca trate este trastorno con un fármaco que la FDA no ha aprobado para tal fin.
Salud con cosas. Ejercicio físico: prescripción en la consulta y cambio social
Los tiempos
cambian, al estilo Dylan, y se nota. Un claro ejemplo es el ejercicio
físico, sea en gimnasios, o corriendo o saliendo a caminar. Municipios
que adaptan zonas para correr, caminar o hacer bici (ruta del
colesterol), zonas con máquinas sencillas de estiramiento para gente
mayor, apps para monitorizar la actividad, juegos para incentivar a
pequeños y mayores... Todo vale, y se nota. Pero hace falta más...
En un reciente editorial del BMJ, titulado "Exercise: not a miracle cure, just good medicine", se comenta un reciente informe
de la Academy of Medical Royal Colleges sobre la importancia del
ejercicio físico para la salud de la población. Pero claro, ¿qué
capacidad de persuasión tienen los profesionales sanitarios? ¿Cuantos
pacientes empiezan a hacer ejercicio tras recibir un consejo de su
profesional de cabecera? Además, no es oro todo lo que parece...
Tal y como
cuentan en el BMJ, la promoción de ejercicio no es cosa de la consulta,
ya que se trata de un cambio multisectorial: educativo, urbanístico,
social, etc. Propuestas como Por un millón de pasos
en Andalucía, o como los grupos de paseo en muchos centros de salud,
son necesarias y, curiosamente, su coste es muy bajo. Quizás sea el
momento de empezar a compartir experiencias e ideas sobre la promoción
del ejercicio dentro y fuera de la consulta. Y también de recopilar
guías (como esta de prescripción de ejercicio de la Sociedad Española de Hipertensión), documentos, vídeos, etc. Además, webs como la de Papps y faecap cuentan con recursos e información, pero hay que ir más allá.
En España, en diciembre de 2013 se aprobó la Estrategia de promoción de la salud y prevención en el SNS. Ahora, como siempre, a saltar de la teoría a la práctica... Quizás haya que volver a decir aquello de "salud en todas las políticas".

viernes, 27 de marzo de 2015
jueves, 26 de marzo de 2015
Docencia Alto Palancia. Página de Cuidados Paliativos
Os presentamos la actualización de la pestaña de Cuidados Paliativos donde encontraréis todas las sesiones que tienen relación con los Cuidados Paliativos: herramientas, aspectos legales y éticos, el manejo de los fármacos más comunes, la rotación de opioides, el duelo o el cine.
http://docenciaaltopalancia.blogspot.com.es/2015/03/pagina-de-cuidados-paliativos.html

miércoles, 25 de marzo de 2015
martes, 24 de marzo de 2015
Viletanos. Inflamación Glándulas Salivares.
Con el título de Inflamación de las Glándulas Salivares, Marina Grueso
(R3 MFyC) presenta una sesión el la que repasa las patologías más
prevalentes que afectan a estas glándulas exocrinas. Describirá procesos
producidos por infecciones bacterianas y víricas, la sialodenitis
necrotizante subaguda, la sialodenitis crónica y hará un repaso sobre
criterios de malignidad.
Puntos Clave:
Puntos Clave:
- Las infecciones bacterians agudas o crónicas tienen un amplo espectro de severidad, en parte debido a la patología previa del paciente.
- La realización de una buena historia clínica indagando sobre los síntomas e ingesta de medicamentos nos puede orientar sobre la etiología.
- El tratamiento suele ser conservador y se puede realizar desde atención primaria.
- Derivar a ORL los casos crónicos o sospecha de neoplasias.
- Ospina A, del Valle F, Naranjo R. Inflamación de Glándulas Salivares , Revisión Bibliográfica. Universidad de Antioquía. revista de Odontología. Volum 15. Num 1 (2004). [Citado el 18-3-2015]. Disponible en internet en: http://aprendeenlinea.udea.edu.co/revistas/index.php/odont/article/view/2496
- Gonzalez FB, García I, García JE. Absceso Parotídeo Bacteriano. Enferm Infecc Microbiol Clin 2008;26(1):65-6. [Citado el 18-3-2015]. Disponible en internet en: http://apps.elsevier.es/watermark/ctl_servlet?_f=10&pident_articulo=13114402&pident_usuario=0&pcontactid=&pident_revista=28&ty=70&accion=L&origen=zonadelectura&web=www.elsevier.es&lan=es&fichero=28v26n01a13114402pdf001.pdf
lunes, 23 de marzo de 2015
3 clics. ¿Cuál es la mejor gliptina?
Comptagotes (18/03/2015)
Joan Antoni Vallès
La comercialización de la sitagliptina en 2006 aportó un nuevo mecanismo de acción -la inhibición de la enzima dipeptil-petidasa-4 (DPP4) - en el tratamiento de la diabetes. Posteriormente, han surgido otros como la vildagliptina (Vilda), saxagliptina (Saxa) linagliptina (lina). Para poder recomendarlos, estos fármacos deberían responder a las necesidades de control glucémico, disminuir las complicaciones micro y macrovasculares, tener un buen perfil de seguridad, ser una buena opción en poblaciones especiales y facilitar la adherencia del tratamiento.
Las indicaciones de todas las gliptinas son muy parecidos: monoterapia, biterapia (junto con metformina, sulfonilureas, pioglitazona o insulina) y en terapia triple.
En la farmacocinética, hay diferencias de biodisponibilidad absoluta entre lina (30%), Saxa (75%), Vilda (85%) y sita (87%). Los tiempos hasta la máxima concentración plasmática son similares (entre 1 y 4 horas). La vida media plasmática terminal también presenta diferencias: desde la Vilda (2-3 horas pero con una lenta disociación de la enzima) en la Saxa (6 horas contando la del metabolito principal), sita (10-12 h) y lina (12 h).
El efecto inhibitorio competitivo en la actividad plasmática de la DPP4 a las 24 horas es diferente entre Vilda (35%), Saxa (69%) y sita o lina (> 80%), lo que se relaciona con mantener el efecto terapéutico en caso de olvido de dosis o mal cumplimiento.
En cuanto a la selectividad hacia los DPP4 en comparación a la actividad en DPP-8 o DPP-9 (la estimulación de estos dos podría asociarse a efectos sobre células de la sangre, sobre la inmunidad, presencia de gammapatías , etc.), la Vilda es entre 32 y 250 veces más selectiva para DPP4 que para DPP8 / DPP, Saxa entre 75 y 400 veces, sita unas 2,600 veces y lina> 10.000 veces.
En cuanto a las vías de metabolización y excreción,
1. Vilda elimina por hidrólisis hepática (69%) y renal o intestinal en menor proporción. No requiere ajuste de dosis en la insuficiencia renal leve o moderada pero, en cambio, no se recomienda su uso en insuficiencia renal aguda o hemodiálisis (ya que aumenta la concentración de su metabolito).
2. Saxa es la única gliptina que se metaboliza por el citocromo P450 hepático y genera un metabolito activo que consigue una vida media de 3,1 horas. Por eso no se puede prescribir en insuficiencia hepática grave. En parte (24% -36%), la Saxa y su metabolito se excretan inalterados por orina.
3. Sita: casi el 80% de la dosis se excreta por la orina sin cambios. En pacientes con un filtrado glomerular entre 30 y 50 ml / min se puede recomendar una dosis segura de 50 mg / día (y de 25 mg / día si el filtrado es inferior a 30 ml / min).
4. Lina: sólo el 7% de la dosis se excreta por riñón. Es una gliptina segura en pacientes con insuficiencia renal.
En resumen, sitagliptina y linagliptina tienen unas características farmacocinéticas interesantes: mayor biodisponibilidad absoluta, vida media plasmática terminal, efecto inhibitorio competitivo sobre la actividad plasmática de la DPP4 a las 24 horas, selectividad hacia los DPP4 en comparación a la actividad en DPP8 o DPP9.
Reducción de la HbA1c. Los mayores niveles de hormonas incretinas, el aumento de la captación tisular de glucosa y de la secreción de la insulina postprandial y la reducción de la secreción de glucagón del páncreas se asocia a una reducción de la HbA1c de entre 0, 3-0,5% en pacientes caucasianos (0,6-0,8% en el conjunto de pacientes incluidos en los ensayos clínicos).
Seguridad. Las incidencias de efectos adversos suelen estar en torno al 10%. Las tasas de abandono por efectos adversos asociados a las gliptinas oscilan entre 2 y 5%, sin claras diferencias entre ellas.
Hay efectos adversos que se han descrito para todas las gliptinas: dolor de cabeza, infecciones (nasofaringitis, gripe, bronquitis, sinusitis, urinarias y dentales), locomotores (artralgia, osteoartritis, dolor de espalda), cutáneos (edema, angioedema, erupciones, dermatitis de contacto, hipersensibilidad, anafilaxia, urticaria) y digestivos (pancreatitis, diarrea, estreñimiento, dolor abdominal, dispepsia).
Hay efectos adversos específicos de la Vilda: hepatitis y otras disfunciones hepáticas (con elevación de las enzimas hepáticas). Por ello, la Vilda debe iniciarse con 50 mg / día y requiere determinar las concentraciones enzimáticas en todos los pacientes cada tres meses el primer año y periódicamente después; hay que suspenderla en caso de hepatitis o ictericia. Otros efectos específicos de la Vilda son alteraciones del sistema nervioso y trastornos de la conducción cardiaca. No se recomienda su uso en pacientes con ICC NYHA III-IV. En relación a la Saxa, hay una nota informativa dirigida a los profesionales (2012) sobre los riesgos de pancreatitis y de reacciones de hipersensibilidad grave asociadas a la saxagliptina.
No hay diferencias significativas en el riesgo cardiovascular entre Saxa o lina frente a placebo. Sin embargo, el estudio SAVOR-TIMI concluyó que saxagliptina se asoció con un aumento del riesgo de hospitalización por insuficiencia cardiaca, más elevado entre los pacientes con niveles elevados de péptidos natriuréticos, insuficiencia cardíaca previa o enfermedad renal crónica.
La seguridad a largo plazo de todas las gliptinas es desconocida, especialmente en cuanto a posibles trastornos del sistema inmunitario.
Repercusiones en la práctica clínica
Las gliptinas, pese a no haber sido comparadas entre ellas, muestran bastante parecidos en cuanto a eficacia y algunas diferencias en cuanto a seguridad. Su eficacia es muy modesta en variables intermedias como la reducción de la HbA1c y no han demostrado beneficios en cuanto a las complicaciones micro o macrovasculares de la diabetes. Se han asociado a efectos adversos como procesos infecciosos, trastornos articulares, cutáneos y digestivos, algunos de los cuales graves. Quizás por todo ello, su uso es prácticamente nulo en algunos países europeos (Holanda, por ejemplo).
El uso en terapéutica de las gliptinas debería ser residual, reservado a los pacientes que necesitan triple terapia oral, recordando que cuando hay que añadir un tercer fármaco la primera opción a valorar debería ser la insulina.
Sitagliptina es la que se ha asociado a menos problemas de seguridad, dispone de una mayor experiencia de uso e incluso se puede usar -Ajuste las dosis- en pacientes con insuficiencia renal.
Joan Antoni Vallès
La comercialización de la sitagliptina en 2006 aportó un nuevo mecanismo de acción -la inhibición de la enzima dipeptil-petidasa-4 (DPP4) - en el tratamiento de la diabetes. Posteriormente, han surgido otros como la vildagliptina (Vilda), saxagliptina (Saxa) linagliptina (lina). Para poder recomendarlos, estos fármacos deberían responder a las necesidades de control glucémico, disminuir las complicaciones micro y macrovasculares, tener un buen perfil de seguridad, ser una buena opción en poblaciones especiales y facilitar la adherencia del tratamiento.
Las indicaciones de todas las gliptinas son muy parecidos: monoterapia, biterapia (junto con metformina, sulfonilureas, pioglitazona o insulina) y en terapia triple.
En la farmacocinética, hay diferencias de biodisponibilidad absoluta entre lina (30%), Saxa (75%), Vilda (85%) y sita (87%). Los tiempos hasta la máxima concentración plasmática son similares (entre 1 y 4 horas). La vida media plasmática terminal también presenta diferencias: desde la Vilda (2-3 horas pero con una lenta disociación de la enzima) en la Saxa (6 horas contando la del metabolito principal), sita (10-12 h) y lina (12 h).
El efecto inhibitorio competitivo en la actividad plasmática de la DPP4 a las 24 horas es diferente entre Vilda (35%), Saxa (69%) y sita o lina (> 80%), lo que se relaciona con mantener el efecto terapéutico en caso de olvido de dosis o mal cumplimiento.
En cuanto a la selectividad hacia los DPP4 en comparación a la actividad en DPP-8 o DPP-9 (la estimulación de estos dos podría asociarse a efectos sobre células de la sangre, sobre la inmunidad, presencia de gammapatías , etc.), la Vilda es entre 32 y 250 veces más selectiva para DPP4 que para DPP8 / DPP, Saxa entre 75 y 400 veces, sita unas 2,600 veces y lina> 10.000 veces.
En cuanto a las vías de metabolización y excreción,
1. Vilda elimina por hidrólisis hepática (69%) y renal o intestinal en menor proporción. No requiere ajuste de dosis en la insuficiencia renal leve o moderada pero, en cambio, no se recomienda su uso en insuficiencia renal aguda o hemodiálisis (ya que aumenta la concentración de su metabolito).
2. Saxa es la única gliptina que se metaboliza por el citocromo P450 hepático y genera un metabolito activo que consigue una vida media de 3,1 horas. Por eso no se puede prescribir en insuficiencia hepática grave. En parte (24% -36%), la Saxa y su metabolito se excretan inalterados por orina.
3. Sita: casi el 80% de la dosis se excreta por la orina sin cambios. En pacientes con un filtrado glomerular entre 30 y 50 ml / min se puede recomendar una dosis segura de 50 mg / día (y de 25 mg / día si el filtrado es inferior a 30 ml / min).
4. Lina: sólo el 7% de la dosis se excreta por riñón. Es una gliptina segura en pacientes con insuficiencia renal.
En resumen, sitagliptina y linagliptina tienen unas características farmacocinéticas interesantes: mayor biodisponibilidad absoluta, vida media plasmática terminal, efecto inhibitorio competitivo sobre la actividad plasmática de la DPP4 a las 24 horas, selectividad hacia los DPP4 en comparación a la actividad en DPP8 o DPP9.
Reducción de la HbA1c. Los mayores niveles de hormonas incretinas, el aumento de la captación tisular de glucosa y de la secreción de la insulina postprandial y la reducción de la secreción de glucagón del páncreas se asocia a una reducción de la HbA1c de entre 0, 3-0,5% en pacientes caucasianos (0,6-0,8% en el conjunto de pacientes incluidos en los ensayos clínicos).
Seguridad. Las incidencias de efectos adversos suelen estar en torno al 10%. Las tasas de abandono por efectos adversos asociados a las gliptinas oscilan entre 2 y 5%, sin claras diferencias entre ellas.
Hay efectos adversos que se han descrito para todas las gliptinas: dolor de cabeza, infecciones (nasofaringitis, gripe, bronquitis, sinusitis, urinarias y dentales), locomotores (artralgia, osteoartritis, dolor de espalda), cutáneos (edema, angioedema, erupciones, dermatitis de contacto, hipersensibilidad, anafilaxia, urticaria) y digestivos (pancreatitis, diarrea, estreñimiento, dolor abdominal, dispepsia).
Hay efectos adversos específicos de la Vilda: hepatitis y otras disfunciones hepáticas (con elevación de las enzimas hepáticas). Por ello, la Vilda debe iniciarse con 50 mg / día y requiere determinar las concentraciones enzimáticas en todos los pacientes cada tres meses el primer año y periódicamente después; hay que suspenderla en caso de hepatitis o ictericia. Otros efectos específicos de la Vilda son alteraciones del sistema nervioso y trastornos de la conducción cardiaca. No se recomienda su uso en pacientes con ICC NYHA III-IV. En relación a la Saxa, hay una nota informativa dirigida a los profesionales (2012) sobre los riesgos de pancreatitis y de reacciones de hipersensibilidad grave asociadas a la saxagliptina.
No hay diferencias significativas en el riesgo cardiovascular entre Saxa o lina frente a placebo. Sin embargo, el estudio SAVOR-TIMI concluyó que saxagliptina se asoció con un aumento del riesgo de hospitalización por insuficiencia cardiaca, más elevado entre los pacientes con niveles elevados de péptidos natriuréticos, insuficiencia cardíaca previa o enfermedad renal crónica.
La seguridad a largo plazo de todas las gliptinas es desconocida, especialmente en cuanto a posibles trastornos del sistema inmunitario.
Repercusiones en la práctica clínica
Las gliptinas, pese a no haber sido comparadas entre ellas, muestran bastante parecidos en cuanto a eficacia y algunas diferencias en cuanto a seguridad. Su eficacia es muy modesta en variables intermedias como la reducción de la HbA1c y no han demostrado beneficios en cuanto a las complicaciones micro o macrovasculares de la diabetes. Se han asociado a efectos adversos como procesos infecciosos, trastornos articulares, cutáneos y digestivos, algunos de los cuales graves. Quizás por todo ello, su uso es prácticamente nulo en algunos países europeos (Holanda, por ejemplo).
El uso en terapéutica de las gliptinas debería ser residual, reservado a los pacientes que necesitan triple terapia oral, recordando que cuando hay que añadir un tercer fármaco la primera opción a valorar debería ser la insulina.
Sitagliptina es la que se ha asociado a menos problemas de seguridad, dispone de una mayor experiencia de uso e incluso se puede usar -Ajuste las dosis- en pacientes con insuficiencia renal.
B MJ. La rosuvastatina no esta en su mejor momento
– La rosuvastatina no esta en su mejor momento. En el último número del BMJ se publicaba un duro artículo sobre
su pobre nivel de evidencia y sus elevadas ventas. Un claro ejemplo de
cómo el marketing sigue siendo crucial para el mundo farmacéutico. Para
saber algo más, no os perdáis esta reciente entrada en el Rincón de Sísifo.
sábado, 21 de marzo de 2015
Viletanos. Vía Aérea.
Manel Carro y Antonia Roca
(Médicos de Familia), tomando como base material de la SeMicyuc sobre
el Plan Nacional de RCP, nos prepararon un taller sobre Vía Aéra,
explicando la técnica para realizar
la intubación traqueal y para la colocación de la mascarilla laríngea. Empleamos dos maniquíes de RCP para practicar dichas técnicas.

Puntos Clave:
Enlaces de Interés:
http://viletanos.blogspot.com.es/2015/03/100310-via-aerea.html?utm_source=feedburner&utm_medium=email&utm_campaign=Feed:+Viletanos+%28VILETANOS%29
la intubación traqueal y para la colocación de la mascarilla laríngea. Empleamos dos maniquíes de RCP para practicar dichas técnicas.
Puntos Clave:
- La intubación traqueal es el método de elección definitivo para el aislamiento de la vía aérea.
- Permite sellar y proteger contra el paso de cuerpos extraños, facilita la ventilación y la aspiración de secreciones y es una vía alternativa para la administración de drogas.
- Cada intento de intubación no debe de superar los 30 segundos de duración. Con el paciente intubado no es necesario mantener la sincronización entre ventilacion y masaje.
- La mascarilla laríngea es un dispositivo alternativo de intubación que permite lograr el aislamiento de la vía aérea con mayor sencillez. Su colocación se hace sin necesidad de visualizar directamente la laringe.
Enlaces de Interés:
- Sociedad Española de Medicina Intensiva, Crítica y Unidades Coronarias (SEMICYUC). http://www.semicyuc.org/temas/
semicyuc/documentos/documento- oficial-de-la-semicyuc/ semicyuc
http://viletanos.blogspot.com.es/2015/03/100310-via-aerea.html?utm_source=feedburner&utm_medium=email&utm_campaign=Feed:+Viletanos+%28VILETANOS%29
viernes, 20 de marzo de 2015
IntraMed. ¿Qué son las α-talasemias?
Autor: Frédéric B. Piel, David J. Weatherall N Engl J Med 2014; 371: 1908-1916. nejm.org DOI: 10.1056/NEJMra1404415. The α-Thalassemias
Introducción
A nivel mundial, las formas más importantes son la α- y la
β-talasemia, que afectan a la producción de las cadenas de α-globina y
β-globina, respectivamente. Aunque la β-talasemia es la forma clínicamente más significativa, la α-talasemia
se produce con una alta frecuencia a lo largo de la zona tropical, casi
llegando a la fijación (un término de la genética de poblaciones que
denota que un alelo mutante de un gen particular se ha convertido en el
único alelo expresado en la población - es decir, que ha llegado a una
frecuencia del 100%) en partes del sur de Asia. Se ha estimado que
alrededor del 5% de la población en todo el mundo porta una variante de
α-talasemia.
Hay evidencia creciente de que la carga sanitaria y económica de las talasemias es cada vez mayor debido al crecimiento de la población, a la transición epidemiológica en regiones tropicales y a las migraciones humanas en otras partes del mundo. (La transición epidemiológica se refiere a un cambio en los patrones de distribución de las edades, la mortalidad, la fecundidad, la esperanza de vida, y las causas de muerte de la población, por lo general reflejado por un cambio de las muertes causadas por enfermedades infecciosas a las muertes causadas por enfermedades crónicas y degenerativas).
El crecimiento de la población conduce a un aumento absoluto del número de nacidos afectados. La transición epidemiológica mejora el diagnóstico de las hemoglobinopatías y la sobrevida de las personas afectadas, aumentando la incidencia de los trastornos. Las migraciones de la población, aunque no siempre conducen a un aumento de la prevalencia global, contribuyen a la propagación de las hemoglobinopatías aumentando por lo tanto el número de países que requieren la aplicación de intervenciones específicas para educar a poblaciones más grandes, diagnosticar los trastornos, y asesorar a los pacientes afectados teniendo en cuenta estas intervenciones en su presupuesto de salud.
Aunque el conocimiento epidemiológico de la distribución, la prevalencia, la genética, la diversidad, y la carga sanitaria de la α-talasemia y la β-talasemia es limitado y en gran medida obsoleto, las brechas son más pronunciadas en el caso de la α-talasemia. Esta relativa falta de una base sólida de pruebas es probable que contribuya a la baja prioridad de este trastorno en muchas agendas de salud pública. Las revisiones existentes se centran principalmente en los aspectos moleculares y clínicos de la α-talasemia.
El objetivo de este artículo es proporcionar un resumen contemporáneo del conocimiento epidemiológico sobre la α-talasemia y discutir los diversos retos que enfrentan las comunidades médicas y de salud pública a la luz de los recientes descubrimientos sobre la gravedad y la genética de este trastorno heredado.
Relevancia Clínica
La hemoglobina adulta normal consiste en pares de cadenas α y β (α2β2), y la hemoglobina fetal tiene dos cadenas α y dos cadenas γ (α2γ2). Los genes para las cadenas α y las cadenas γ están duplicados (αα/αα, γγ/γγ), mientras que las cadenas β son codificadas por un locus único (β/β).
En el feto, la producción defectuosa de las cadenas α se refleja por la presencia de cadenas γ en exceso, que forman un tetrámero γ4, llamado hemoglobina de Bart; en los adultos, el exceso de cadenas β forman un tetrámero β4, llamado hemoglobina H (HbH). Debido a su muy alta afinidad por el oxígeno, ambos tetrámeros no pueden transportar oxígeno, y, en el caso de la HbH, su inestabilidad conduce a la producción de cuerpos de inclusión en los glóbulos rojos y a un grado variable de anemia hemolítica.
Hasta el momento se han identificado más de 100 formas genéticas de α-talasemia, con fenotipos que varían de asintomáticos a letales. A pesar de esta complejidad, la gravedad de este trastorno por lo general se correlaciona bien con el número de copias no funcionales de los genes de α-globina.
En base al número de genes de α-globina perdidos por deleción o inactivados total o parcialmente por mutaciones puntuales, las α-talasemias se clasifican en dos subgrupos principales: α+-talasemia (anteriormente llamada α-talasemia 2), en la que un par de los genes es eliminado o inactivado por una mutación puntual (-α/αα o ααND/αα, con ND denotando no deleción), y α0-talasemia (anteriormente llamada α-talasemia 1), en la que ambos pares de genes α-globina en el mismo cromosoma son suprimidos (--/αα).
Las formas clínicamente relevantes de α-talasemia por lo general implican a la α0-talasemia, ya sea co-heredada con α+-talasemia (-α/-- o ααND/--) resultando en enfermedad de HbH o heredada de ambos padres y resultando en hidropesía fetal por hemoglobina de Bart (--/--), que es letal en el útero o poco después del nacimiento. Los embriones afectados sucumben a la hipoxia severa ya sea temprano en la gestación (por ejemplo, en el caso de --FIL/--FIL [donde FIL se refiere a una deleción que causa α0-talasemia y que es prevalente entre los filipinos]) o durante el tercer trimestre (por ejemplo, en el caso de --SEA/--SEA [donde SEA se refiere a una deleción que causa α0-talasemia y que es frecuente entre personas del sudeste asiático]).
Pocos niños con hidropesía fetal por hemoglobina de Bart que recibieron una transfusión intrauterina o una transfusión inmediatamente después del parto han sobrevivido hasta los 5 años de edad. Estos niños requieren transfusiones periódicas y, cuando es apropiado, terapia de quelación del hierro; ellos por lo general tienen graves complicaciones clínicas, anomalías congénitas, y retrasos en las funciones cognitivas y motoras.
El síndrome de hidropesía fetal por hemoglobina de Bart a menudo se acompaña por una variedad de malformaciones congénitas y complicaciones maternas, incluyendo anemia severa del embarazo, preeclampsia, polihidramnios, y dificultades graves para expulsar el feto y la placenta sumamente agrandada. Aunque estas complicaciones han sido bien documentadas, hay datos muy limitados en cuanto a la frecuencia de las muertes maternas, sobre todo en los países en desarrollo en los que esta condición es tan común.
La enfermedad por HbH es considerada a menudo un trastorno relativamente leve. Sin embargo, los estudios han destacado ciertos fenotipos clínicamente graves, en particular en variantes no delecionales de la enfermedad. De hecho, la enfermedad por HbH se caracteriza por una amplia gama de características fenotípicas. La forma que resulta de deleciones (-α/--) por lo general sigue un curso relativamente leve, con anemia moderada y esplenomegalia.
Aparte de los episodios de infección intercurrente, esta forma de enfermedad por HbH no requiere transfusiones de sangre. Sin embargo, la variedad que resulta de la interacción de una mutación del gen α-globina no delecional junto con α0-talasemia (ααND/--) sigue un curso mucho más severo. Esto es particularmente cierto cuando la mutación no delecional es en la terminación de la cadena de α-globina de la hemoglobina mutante Constant Spring, que es muy común en muchos países asiáticos.
Las formas no delecionales de la enfermedad por HbH se caracterizan por una anemia grave, que ocurre a menudo en la vida temprana, y se asocian con esplenomegalia en aumento, una carga importante de hierro, y una variedad de otras complicaciones clínicas, incluyendo infecciones, úlceras en las piernas, cálculos biliares, y deficiencia de ácido fólico. Aunque en general está indicada la esplenectomía, la enfermedad por HbH no delecional se asocia con una tasa particularmente alta de complicaciones trombóticas. Esta observación hace que la decisión entre la esplenectomía y la transfusión de por vida sea extremadamente difícil.
Variantes más leves de α-talasemia actúan como modificadores genéticos de otras enfermedades hereditarias, como se ilustra por las interacciones epistáticas (cuando un gen influye en otro) entre la α-talasemia y la β-talasemia o entre la α-talasemia y la hemoglobina S (hemoglobina falciforme). Se han observado frecuentemente triplicaciones y cuadruplicaciones del gen de la α-globina en muchas poblaciones, y pueden interactuar con las variantes de β-talasemia para producir fenotipos más graves.
Por último, hay dos síndromes en los que la α-talasemia se asocia con retraso mental (Síndromes ATR). Los detalles relativos a estos síndromes se proporcionan en el Apéndice Suplementario, disponible con el texto completo de este artículo en NEJM.org.
Diagnóstico
Se requiere un diagnóstico prenatal para identificar a los fetos afectados por hidropesía fetal secundaria a hemoglobina de Bart y para reducir los riesgos para las madres. La decisión de considerar este diagnóstico por lo general lleva al hallazgo de glóbulos rojos microcíticos hipocrómicos en ambos padres, en asociación con un nivel normal de hemoglobina A2; esta combinación descartaría la β-talasemia, que por lo general implica un nivel elevado de hemoglobina A2. La deficiencia de hierro también tiene que ser descartada.
Cuando se dispone de instalaciones para el diagnóstico rápido de ADN, el examen hematológico es seguido por la confirmación de la presencia de α0-talasemia en los padres. El diagnóstico fetal por lo general se hace al principio del embarazo mediante una muestra de vellosidades coriónicas, aunque la anemia fetal también puede diagnosticarse después durante la gestación por cuantificación de la velocidad sistólica pico en la arteria cerebral media.
Varios métodos alternativos de diagnóstico genético pre-implantación y preconcepción o de diagnóstico prenatal - por ejemplo, análisis de ADN fetal en sangre materna e identificación de células fetales en sangre materna por tinción con anticuerpos contra las cadenas de globina - todavía están en etapas relativamente tempranas de estudio. Mientras tanto, los intentos de tratamiento intrauterino y postnatal se asocian con numerosos desafíos éticos.
El estado homocigoto de α+-talasemia y el estado heterocigoto de α0-talasemia (agrupados bajo el término "α-talasemia menor") se asocian con una reducción sustancial del volumen corpuscular medio (VCM) y de la hemoglobina corpuscular media (HCM). En los heterocigotos para α+-talasemia, el VCM y la HCM en general están disminuidos, pero hay una pequeña superposición con los valores normales. Las formas más leves de α-talasemia se diagnostican a menudo como deficiencia de hierro, aunque la frecuencia exacta de este diagnóstico erróneo es desconocida. En última instancia, el diagnóstico de una variante particular de la α-talasemia puede confirmarse sólo a nivel del ADN.
En la era pre-genómica, la frecuencia de α-talasemia se evaluó en base a la presencia de hemoglobina de Bart en sangre de cordón. La detección de hemoglobina de Bart en los recién nacidos indica que uno o más de los cuatro genes de α-globina son disfuncionales, causando α-talasemia. Aunque se pensó inicialmente que el nivel de hemoglobina de Bart al nacer sería un indicador sensible de la presencia de α-talasemia y que se correlacionaría bien con su gravedad, los estudios basados en ADN posteriores mostraron que este método diagnóstico falla en detectar un número sustancial de casos heterocigotos de α+-talasemia y, por lo tanto subestima la frecuencia de α-talasemia.
Actualmente está bien establecido que el diagnóstico de α-talasemia en base a la hemoglobina de Bart por sí sola no es confiable y no permite la identificación de los genotipos. Este método, sin embargo, sigue siendo ampliamente utilizado en países de bajos y medianos ingresos, porque es relativamente sencillo y mucho más barato que el análisis del ADN.
Distribución geográfica
La evidencia de que la α-talasemia es altamente protectora contra la malaria severa está bien establecida. Como resultado de esta ventaja selectiva, la α-talasemia heterocigota ha alcanzado altas frecuencias en todas las regiones tropicales y subtropicales, incluyendo la mayor parte del sudeste de Asia, el área del Mediterráneo, el subcontinente Indio, Oriente Medio y África.
Variantes comunes de α0-talasemia, predominantemente la mutación --SEA en el sudeste de Asia y la mutación --MED en el Mediterráneo, han llegado a frecuencias de aproximadamente 5%. Aunque hay por lo menos siete formas delecionales de α+-talasemia, las variantes -α3.7 son las más comunes. Se han reportado frecuencias del 70% y de hasta el 90% en Melanesia y en partes de Nepal, respectivamente.
Los mecanismos por los que se han alcanzado tales frecuencias cercanas a la fijación requieren mayor investigación. Además de los estudios que revelaron epistaxis negativa entre los pacientes con α+-talasemia y rasgo de células falciformes, resultando en un nivel reducido de protección contra la malaria cuando los dos son coheredados, modelos matemáticos han sugerido que la frecuencia de α+-talasemia podría estar limitada por la presencia del gen falciforme en África y el Mediterráneo.
En conjunción con los movimientos poblacionales globales a gran escala en las últimas décadas, la α-talasemia se ha extendido a muchas otras partes del mundo, incluyendo el norte de Europa y el Norte de América. Este fenómeno está mejor ilustrado por la aplicación en 1998 de un programa de cribado universal para α-talasemia en California. Después de la inmigración de un gran número de personas de Filipinas y de otros países del sudeste asiático, la incidencia de síndromes de α-talasemia en California entre enero de1998 y junio de 2006 fue de 11,1 casos por cada 100.000 personas evaluadas, con 406 casos de enfermedad por HbH y 5 casos de hidropesía fetal por hemoglobina de Bart.
Debido a la alta frecuencia de variantes de α+-talasemia en todo el mundo, es probable que, con la mezcla de poblaciones locales e inmigrantes, tal propagación aumente la incidencia de enfermedad por HbH, además de crear una carga de salud en un número creciente de países o regiones.
Varias encuestas de población sobre hemoglobinopatías revelaron heterogeneidades geográficas notables en la prevalencia de estos desórdenes. A mediados de la década de 1980, Flint y colegas observaron frecuencias de α+-talasemia que variaban del 6 al 68% a lo largo de las islas melanesias. Se describieron heterogeneidades similares más tarde en Vanuatu.
En una encuesta de micro-mapeo reciente llevada a cabo en Sri Lanka, la prevalencia de α-talasemia osciló entre el 2 y el 20% (Weatherall DJ, y col.: datos inéditos). Esta variabilidad es probable que sea el resultado de una serie de factores ambientales complejos, incluyendo la tasa de transmisión de la malaria. Una mejor comprensión de estas interacciones ayudará considerablemente en la refinación de las estimaciones de las poblaciones afectadas en las regiones tropicales y en las políticas de desarrollo para el diagnóstico y manejo de la α-talasemia en base a las características de la población local.
Es necesaria una cuantificación precisa de las poblaciones en riesgo de desarrollar síndromes de α-talasemia a nivel nacional, regional, y mundial para definir recursos actuales y futuros requeridos para proporcionar un diagnóstico prenatal preciso de variantes de α-talasemia, un manejo de emergencia de los embarazos con hidropesía fetal por hemoglobina de Bart y sus complicaciones maternas asociadas, y un manejo a largo plazo de la enfermedad por HbH.
Detección
Los objetivos principales de los programas de detección de talasemia son determinar la frecuencia de las diferentes variantes genéticas observadas en las comunidades e identificar e informar a las parejas que están en riesgo, en particular para las formas graves de la enfermedad presentes en las zonas de alta frecuencia.
La detección de la α-talasemia es especialmente útil en la prevención de complicaciones maternas graves en el caso de hidropesía fetal por hemoglobina de Bart, en la provisión de un diagnóstico preciso en los casos en los que la α-talasemia se cohereda con la hemoglobina S o la β-talasemia, o en casos en los que se detecta deficiencia de hierro. La mayoría de los encuestas de detección de α-talasemia son llevadas a cabo como parte de un programa de prevención de β-talasemia y por lo tanto no son adecuadas para determinar frecuencias de α-talasemia en la población.
Los beneficios del cribado de la población deberán considerarse cuidadosamente en cualquier población en la que las variantes de α-talasemia son frecuentes y cuando se observan casos de anemia microcítica hipocrómica inexplicable en ausencia de deficiencia de hierro.
Carga sanitaria y económica
Los primeros estudios sobre la carga global de los trastornos de la hemoglobina que evaluaron la frecuencia de los mismos y el número de años de vida ajustados por discapacidad (AVAD) asociado con ellos se vieron limitados por la falta de datos sobre la α-talasemia, en particular con respecto a la hidropesía fetal de la hemoglobina de Bart y a su carga en relación con los mortinatos o fallecidos poco después del parto.
La Organización Mundial de la Salud (OMS) no recopila datos sobre los mortinatos, y los únicos datos disponibles proveyeron una estimación de 1.250 embarazos con α0-talasemia homocigota por año en Tailandia, una cifra que corresponde a 37.242 AVAD. En comparación, se estimó que la β-talasemia homocigota y la hemoglobina E/β-talasemia resultaron en un total de 53.600 AVAD en Tailandia, en base a una esperanza de vida de 10 años y 30 años, respectivamente. Estas son probablemente subestimaciones porque se consideró la discapacidad de la madre sólo en el último trimestre del embarazo, sin tener en cuenta las complicaciones postnatales que pueden resultar en muerte, y porque no se calcularon las estimaciones para la enfermedad por HbH, ya que se realizó la recolección de los datos antes de que ocurriera una forma particularmente severa de la enfermedad por HbH en muchos países asiáticos.
La información sobre la incidencia de α-talasemia fue insuficiente para permitir el cálculo de la carga regional y global de enfermedad. Se han reportado recientemente estimaciones globales del número de AVAD resultantes de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad del 2010, aunque aún no se han añadido a este proyecto datos precisos sobre los diversos subtipos de talasemias.
Para el conocimiento de los autores, hay un solo estudio hasta la fecha que ha investigado la relación costo-beneficio de un programa de prevención de α-talasemia, establecido por sí mismo o como parte de un amplio programa para todas las talasemias o para condiciones genéticas (por ejemplo, fenilcetonuria) en general. El estudio, llevado a cabo en Hong Kong, concluyó que el cribado prenatal universal para α- y β-talasemias con el uso de pruebas de ADN era rentable, aunque no se dispuso de datos publicados sobre el costo del manejo de un embarazo afectado por hidropesía fetal secundaria a hemoglobina de Bart.
Debe investigarse cuidadosamente si estos resultados son aplicables a países con mayores poblaciones y menores ingresos que los de Hong Kong. Los beneficios del diagnóstico preciso de la deficiencia de hierro y otras hemoglobinopatías - Hemoglobina S y β-talasemia en particular - también deben ser considerados. Los análisis de costo-beneficio de los programas de prevención de β-talasemia en Quebec, Irán, Israel, y el Reino Unido generalmente han confirmado los beneficios generales de este tipo de programas (es decir, los costos de prevención fueron inferiores a los costos de tratamiento), pero los estudios no incluyeron a la α-talasemia.
En los países de bajos y medianos ingresos, esos programas podrían beneficiarse de las infraestructuras existentes (por ejemplo, los programas de cribado de fenilcetonuria o de deficiencia de glucosa-6-fosfato deshidrogenasa en curso) o podrían resultar en una mejora general del acceso a la atención de salud para las comunidades locales.
Manejo
El conocimiento detallado de la prevalencia de la α-talasemia (incluyendo el estado de portador) y de su diversidad genética es esencial para definir políticas orientadas a la reducción de la carga sanitaria a largo plazo de las hemoglobinopatías, permitiendo el diagnóstico preciso (y por lo tanto evitando investigaciones inexactas y caras), el establecimiento de la verdadera causa de la microcitosis, el asesoramiento genético adecuado, y la asignación de recursos para hacer frente a las emergencias y a las necesidades a largo plazo de los pacientes y sus familiares de una manera rentable. Para lograr estos objetivos, la comunidad médica se enfrenta a varios desafíos importantes, que se resumen en la Tabla 1.
Tabla 1. Principales desafíos asociados con la carga sanitaria en aumento de la α-talasemia y recomendaciones para superarlos
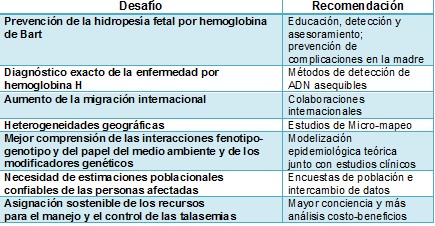 La mayoría de las parejas en riesgo de concebir fetos con hidropesía
fetal por hemoglobina de Bart no son actualmente identificadas. Por lo
tanto, es muy probable que las estimaciones actuales representen grandes
subestimaciones de los mortinatos por este desorden.
La mayoría de las parejas en riesgo de concebir fetos con hidropesía
fetal por hemoglobina de Bart no son actualmente identificadas. Por lo
tanto, es muy probable que las estimaciones actuales representen grandes
subestimaciones de los mortinatos por este desorden.
En ausencia de tratamientos específicos y de una clara comprensión de los mecanismos subyacentes responsables de la amplia gama de anomalías congénitas asociadas con este trastorno letal, el cribado y el diagnóstico prenatal temprano representan las únicas opciones para identificar los embarazos de alto riesgo y evitar complicaciones maternas graves.
Aunque por lo general se recomienda la interrupción de los embarazos afectados debido al aumento del riesgo de complicaciones maternas y fetales severas y a los efectos psicológicos en las familias, deben tenerse en cuenta los antecedentes culturales y religiosos cuando se asesora a las parejas en riesgo, tanto en las comunidades en las que la α-talasemia ha sido prevalente tradicionalmente como en aquellas en las que se ha introducido recientemente a través de la migración.
Un diagnóstico temprano correcto en el embarazo es esencial para evitar complicaciones médicas graves y trauma psicológico. Tal diagnóstico puede hacerse de forma confiable con costos relativamente bajos previendo que las mujeres embarazadas reciban un seguimiento regular por personal médico familiarizado con este síndrome.
Los recientes progresos en las pruebas de ADN revelaron una mayor diversidad fenotípica de la enfermedad por HbH que lo que se pensaba previamente.
Como se ha señalado anteriormente, las variantes no delecionales suelen ser más graves que las variantes de deleción. Por ello es crucial realizar el análisis del ADN para identificar la variante genética subyacente responsable de este trastorno. La reacción en cadena de la polimerasa-Gap (PCR) y los ensayos de PCR multiplex permiten una fácil detección de una gama de variantes comunes, pero tales métodos siguen siendo costosos y no son ampliamente utilizados en países de bajos y medianos ingresos. Con los métodos de prueba de ADN cada vez más y más asequibles, es probable que éstos se conviertan en una parte regular de los servicios disponibles para el control y manejo de las talasemias, en particular en los países asiáticos.
A medida que el conocimiento sobre la relación fenotipo-genotipo en la enfermedad por HbH mejora, la correcta identificación de genotipos será imprescindible para informar a los padres sobre los riesgos reproductivos durante el asesoramiento genético y para dar un adecuado cuidado a los pacientes afectados.
Con el aumento de los movimientos de población, la diversidad de combinaciones de variantes de α-talasemia o hemoglobinopatías coheredadas seguirá aumentando. Aunque es difícil predecir el fenotipo exacto de estas nuevas combinaciones, un conocimiento detallado de la distribución actual de las variantes genéticas ayudará al menos en la definición de los procedimientos de diagnóstico y en la evaluación de los riesgos potenciales.
Las colaboraciones entre las Áreas de "Origen" (aquellas con una alta prevalencia de α-talasemia y emigración sustancial) y las Áreas "Sumidero" (las que tienen inmigración sustancial de las zonas de origen), como las desarrolladas entre Filipinas y California y entre Sri Lanka y el Reino Unido, sin duda representan modelos beneficiosos para ambas partes y deben ser replicados más ampliamente.
La carga sanitaria de la α+-talasemia actualmente no se conoce. Aunque las variantes de α+-talasemia por si solas no representan un problema clínico directo, son probablemente los trastornos genéticos más comunes en el mundo, y representan importantes modificadores genéticos para una serie de condiciones, incluyendo la malaria, los trastornos de células falciformes, la β-talasemia, y la deficiencia de hierro.
Una mejor comprensión de estas interacciones es crucial para proporcionar un diagnóstico y tratamiento adecuado para los pacientes afectados, pero será relevante sólo si las intervenciones resultantes de esta mejor comprensión se implementan en todas las áreas en las que estas interacciones se están produciendo. Por ejemplo, se desconoce actualmente si las interacciones epistáticas entre el rasgo de células falciformes y la α+-talasemia observadas en Kenia también se están produciendo en las poblaciones indígenas.
Debido a la notable heterogeneidad geográfica en la prevalencia de la α-talasemia, las intervenciones tienen que adaptarse a las características específicas de la población local (por ejemplo, prevalencia del trastorno en la población, composición étnica, y consanguinidad) y al sistema de salud local. Una mejor comprensión de la relación entre la prevalencia de las variantes de α-talasemia, los factores ambientales y las infecciones, en combinación con métodos analíticos y de modelización modernos, ayudaría a lograr estimaciones más refinadas de las poblaciones afectadas. Tal conocimiento también ayudaría en el desarrollo de programas de prevención estratificados enfocados, por ejemplo, en regiones en las que las variantes de α0-talasemia son más frecuentes.
Conclusiones
Las α-talasemias representan un problema de salud mundial con una carga creciente. Un conocimiento refinado de las bases moleculares de la α-talasemia será totalmente relevante desde la perspectiva de la salud pública sólo si se complementa con datos epidemiológicos detallados.
Para garantizar el adecuado cuidado de los pacientes y la sostenibilidad de los sistemas de atención de salud, se debe poner mayor esfuerzo en obtener estimaciones basadas en la evidencia de las poblaciones afectadas, la provisión de recursos para la prevención, control y manejo de las talasemias, y realizar análisis de costo-efectividad. Tales objetivos se alcanzarán sólo a través de un esfuerzo concertado de las comunidades de investigación y médicas y el apoyo de agencias de financiación internacional para recopilar y compartir datos epidemiológicos. La reciente inclusión de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad y la evaluación de la carga de la α-talasemia en términos de AVAD tendrán sentido sólo si se dispone de datos confiables y actualizados.
Comentario:
Las talasemias son trastornos hereditarios de la síntesis de la hemoglobina que se caracterizan por una producción reducida de las cadenas de globina. Presentan una amplia distribución mundial debido al movimiento poblacional constante, pero no se cuenta actualmente con datos exactos de prevalencia. Estas migraciones de población contribuyen a la propagación de estas hemoglobinopatías aumentando el número de países que requieren la aplicación de intervenciones específicas para educar a sus comunidades, diagnosticar los trastornos, y asesorar a los pacientes afectados y a sus familias. Se requieren esfuerzos conjuntos para determinar las bases moleculares de las variantes de talasemia y su epidemiología a nivel global y regional, y para desarrollar programas de detección, manejo y asesoramiento con respecto a estos trastornos particulares de la hemoglobina.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol
Introducción
| Las talasemias son las enfermedades humanas monogénicas más comunes. Estos trastornos hereditarios de la síntesis de la hemoglobina se caracterizan por una producción reducida de las cadenas de globina de la hemoglobina. |
Hay evidencia creciente de que la carga sanitaria y económica de las talasemias es cada vez mayor debido al crecimiento de la población, a la transición epidemiológica en regiones tropicales y a las migraciones humanas en otras partes del mundo. (La transición epidemiológica se refiere a un cambio en los patrones de distribución de las edades, la mortalidad, la fecundidad, la esperanza de vida, y las causas de muerte de la población, por lo general reflejado por un cambio de las muertes causadas por enfermedades infecciosas a las muertes causadas por enfermedades crónicas y degenerativas).
El crecimiento de la población conduce a un aumento absoluto del número de nacidos afectados. La transición epidemiológica mejora el diagnóstico de las hemoglobinopatías y la sobrevida de las personas afectadas, aumentando la incidencia de los trastornos. Las migraciones de la población, aunque no siempre conducen a un aumento de la prevalencia global, contribuyen a la propagación de las hemoglobinopatías aumentando por lo tanto el número de países que requieren la aplicación de intervenciones específicas para educar a poblaciones más grandes, diagnosticar los trastornos, y asesorar a los pacientes afectados teniendo en cuenta estas intervenciones en su presupuesto de salud.
Aunque el conocimiento epidemiológico de la distribución, la prevalencia, la genética, la diversidad, y la carga sanitaria de la α-talasemia y la β-talasemia es limitado y en gran medida obsoleto, las brechas son más pronunciadas en el caso de la α-talasemia. Esta relativa falta de una base sólida de pruebas es probable que contribuya a la baja prioridad de este trastorno en muchas agendas de salud pública. Las revisiones existentes se centran principalmente en los aspectos moleculares y clínicos de la α-talasemia.
El objetivo de este artículo es proporcionar un resumen contemporáneo del conocimiento epidemiológico sobre la α-talasemia y discutir los diversos retos que enfrentan las comunidades médicas y de salud pública a la luz de los recientes descubrimientos sobre la gravedad y la genética de este trastorno heredado.
Relevancia Clínica
La hemoglobina adulta normal consiste en pares de cadenas α y β (α2β2), y la hemoglobina fetal tiene dos cadenas α y dos cadenas γ (α2γ2). Los genes para las cadenas α y las cadenas γ están duplicados (αα/αα, γγ/γγ), mientras que las cadenas β son codificadas por un locus único (β/β).
En el feto, la producción defectuosa de las cadenas α se refleja por la presencia de cadenas γ en exceso, que forman un tetrámero γ4, llamado hemoglobina de Bart; en los adultos, el exceso de cadenas β forman un tetrámero β4, llamado hemoglobina H (HbH). Debido a su muy alta afinidad por el oxígeno, ambos tetrámeros no pueden transportar oxígeno, y, en el caso de la HbH, su inestabilidad conduce a la producción de cuerpos de inclusión en los glóbulos rojos y a un grado variable de anemia hemolítica.
Hasta el momento se han identificado más de 100 formas genéticas de α-talasemia, con fenotipos que varían de asintomáticos a letales. A pesar de esta complejidad, la gravedad de este trastorno por lo general se correlaciona bien con el número de copias no funcionales de los genes de α-globina.
En base al número de genes de α-globina perdidos por deleción o inactivados total o parcialmente por mutaciones puntuales, las α-talasemias se clasifican en dos subgrupos principales: α+-talasemia (anteriormente llamada α-talasemia 2), en la que un par de los genes es eliminado o inactivado por una mutación puntual (-α/αα o ααND/αα, con ND denotando no deleción), y α0-talasemia (anteriormente llamada α-talasemia 1), en la que ambos pares de genes α-globina en el mismo cromosoma son suprimidos (--/αα).
Las formas clínicamente relevantes de α-talasemia por lo general implican a la α0-talasemia, ya sea co-heredada con α+-talasemia (-α/-- o ααND/--) resultando en enfermedad de HbH o heredada de ambos padres y resultando en hidropesía fetal por hemoglobina de Bart (--/--), que es letal en el útero o poco después del nacimiento. Los embriones afectados sucumben a la hipoxia severa ya sea temprano en la gestación (por ejemplo, en el caso de --FIL/--FIL [donde FIL se refiere a una deleción que causa α0-talasemia y que es prevalente entre los filipinos]) o durante el tercer trimestre (por ejemplo, en el caso de --SEA/--SEA [donde SEA se refiere a una deleción que causa α0-talasemia y que es frecuente entre personas del sudeste asiático]).
Pocos niños con hidropesía fetal por hemoglobina de Bart que recibieron una transfusión intrauterina o una transfusión inmediatamente después del parto han sobrevivido hasta los 5 años de edad. Estos niños requieren transfusiones periódicas y, cuando es apropiado, terapia de quelación del hierro; ellos por lo general tienen graves complicaciones clínicas, anomalías congénitas, y retrasos en las funciones cognitivas y motoras.
El síndrome de hidropesía fetal por hemoglobina de Bart a menudo se acompaña por una variedad de malformaciones congénitas y complicaciones maternas, incluyendo anemia severa del embarazo, preeclampsia, polihidramnios, y dificultades graves para expulsar el feto y la placenta sumamente agrandada. Aunque estas complicaciones han sido bien documentadas, hay datos muy limitados en cuanto a la frecuencia de las muertes maternas, sobre todo en los países en desarrollo en los que esta condición es tan común.
La enfermedad por HbH es considerada a menudo un trastorno relativamente leve. Sin embargo, los estudios han destacado ciertos fenotipos clínicamente graves, en particular en variantes no delecionales de la enfermedad. De hecho, la enfermedad por HbH se caracteriza por una amplia gama de características fenotípicas. La forma que resulta de deleciones (-α/--) por lo general sigue un curso relativamente leve, con anemia moderada y esplenomegalia.
Aparte de los episodios de infección intercurrente, esta forma de enfermedad por HbH no requiere transfusiones de sangre. Sin embargo, la variedad que resulta de la interacción de una mutación del gen α-globina no delecional junto con α0-talasemia (ααND/--) sigue un curso mucho más severo. Esto es particularmente cierto cuando la mutación no delecional es en la terminación de la cadena de α-globina de la hemoglobina mutante Constant Spring, que es muy común en muchos países asiáticos.
Las formas no delecionales de la enfermedad por HbH se caracterizan por una anemia grave, que ocurre a menudo en la vida temprana, y se asocian con esplenomegalia en aumento, una carga importante de hierro, y una variedad de otras complicaciones clínicas, incluyendo infecciones, úlceras en las piernas, cálculos biliares, y deficiencia de ácido fólico. Aunque en general está indicada la esplenectomía, la enfermedad por HbH no delecional se asocia con una tasa particularmente alta de complicaciones trombóticas. Esta observación hace que la decisión entre la esplenectomía y la transfusión de por vida sea extremadamente difícil.
Variantes más leves de α-talasemia actúan como modificadores genéticos de otras enfermedades hereditarias, como se ilustra por las interacciones epistáticas (cuando un gen influye en otro) entre la α-talasemia y la β-talasemia o entre la α-talasemia y la hemoglobina S (hemoglobina falciforme). Se han observado frecuentemente triplicaciones y cuadruplicaciones del gen de la α-globina en muchas poblaciones, y pueden interactuar con las variantes de β-talasemia para producir fenotipos más graves.
Por último, hay dos síndromes en los que la α-talasemia se asocia con retraso mental (Síndromes ATR). Los detalles relativos a estos síndromes se proporcionan en el Apéndice Suplementario, disponible con el texto completo de este artículo en NEJM.org.
Diagnóstico
Se requiere un diagnóstico prenatal para identificar a los fetos afectados por hidropesía fetal secundaria a hemoglobina de Bart y para reducir los riesgos para las madres. La decisión de considerar este diagnóstico por lo general lleva al hallazgo de glóbulos rojos microcíticos hipocrómicos en ambos padres, en asociación con un nivel normal de hemoglobina A2; esta combinación descartaría la β-talasemia, que por lo general implica un nivel elevado de hemoglobina A2. La deficiencia de hierro también tiene que ser descartada.
Cuando se dispone de instalaciones para el diagnóstico rápido de ADN, el examen hematológico es seguido por la confirmación de la presencia de α0-talasemia en los padres. El diagnóstico fetal por lo general se hace al principio del embarazo mediante una muestra de vellosidades coriónicas, aunque la anemia fetal también puede diagnosticarse después durante la gestación por cuantificación de la velocidad sistólica pico en la arteria cerebral media.
Varios métodos alternativos de diagnóstico genético pre-implantación y preconcepción o de diagnóstico prenatal - por ejemplo, análisis de ADN fetal en sangre materna e identificación de células fetales en sangre materna por tinción con anticuerpos contra las cadenas de globina - todavía están en etapas relativamente tempranas de estudio. Mientras tanto, los intentos de tratamiento intrauterino y postnatal se asocian con numerosos desafíos éticos.
El estado homocigoto de α+-talasemia y el estado heterocigoto de α0-talasemia (agrupados bajo el término "α-talasemia menor") se asocian con una reducción sustancial del volumen corpuscular medio (VCM) y de la hemoglobina corpuscular media (HCM). En los heterocigotos para α+-talasemia, el VCM y la HCM en general están disminuidos, pero hay una pequeña superposición con los valores normales. Las formas más leves de α-talasemia se diagnostican a menudo como deficiencia de hierro, aunque la frecuencia exacta de este diagnóstico erróneo es desconocida. En última instancia, el diagnóstico de una variante particular de la α-talasemia puede confirmarse sólo a nivel del ADN.
En la era pre-genómica, la frecuencia de α-talasemia se evaluó en base a la presencia de hemoglobina de Bart en sangre de cordón. La detección de hemoglobina de Bart en los recién nacidos indica que uno o más de los cuatro genes de α-globina son disfuncionales, causando α-talasemia. Aunque se pensó inicialmente que el nivel de hemoglobina de Bart al nacer sería un indicador sensible de la presencia de α-talasemia y que se correlacionaría bien con su gravedad, los estudios basados en ADN posteriores mostraron que este método diagnóstico falla en detectar un número sustancial de casos heterocigotos de α+-talasemia y, por lo tanto subestima la frecuencia de α-talasemia.
Actualmente está bien establecido que el diagnóstico de α-talasemia en base a la hemoglobina de Bart por sí sola no es confiable y no permite la identificación de los genotipos. Este método, sin embargo, sigue siendo ampliamente utilizado en países de bajos y medianos ingresos, porque es relativamente sencillo y mucho más barato que el análisis del ADN.
Distribución geográfica
La evidencia de que la α-talasemia es altamente protectora contra la malaria severa está bien establecida. Como resultado de esta ventaja selectiva, la α-talasemia heterocigota ha alcanzado altas frecuencias en todas las regiones tropicales y subtropicales, incluyendo la mayor parte del sudeste de Asia, el área del Mediterráneo, el subcontinente Indio, Oriente Medio y África.
Variantes comunes de α0-talasemia, predominantemente la mutación --SEA en el sudeste de Asia y la mutación --MED en el Mediterráneo, han llegado a frecuencias de aproximadamente 5%. Aunque hay por lo menos siete formas delecionales de α+-talasemia, las variantes -α3.7 son las más comunes. Se han reportado frecuencias del 70% y de hasta el 90% en Melanesia y en partes de Nepal, respectivamente.
Los mecanismos por los que se han alcanzado tales frecuencias cercanas a la fijación requieren mayor investigación. Además de los estudios que revelaron epistaxis negativa entre los pacientes con α+-talasemia y rasgo de células falciformes, resultando en un nivel reducido de protección contra la malaria cuando los dos son coheredados, modelos matemáticos han sugerido que la frecuencia de α+-talasemia podría estar limitada por la presencia del gen falciforme en África y el Mediterráneo.
En conjunción con los movimientos poblacionales globales a gran escala en las últimas décadas, la α-talasemia se ha extendido a muchas otras partes del mundo, incluyendo el norte de Europa y el Norte de América. Este fenómeno está mejor ilustrado por la aplicación en 1998 de un programa de cribado universal para α-talasemia en California. Después de la inmigración de un gran número de personas de Filipinas y de otros países del sudeste asiático, la incidencia de síndromes de α-talasemia en California entre enero de1998 y junio de 2006 fue de 11,1 casos por cada 100.000 personas evaluadas, con 406 casos de enfermedad por HbH y 5 casos de hidropesía fetal por hemoglobina de Bart.
Debido a la alta frecuencia de variantes de α+-talasemia en todo el mundo, es probable que, con la mezcla de poblaciones locales e inmigrantes, tal propagación aumente la incidencia de enfermedad por HbH, además de crear una carga de salud en un número creciente de países o regiones.
Varias encuestas de población sobre hemoglobinopatías revelaron heterogeneidades geográficas notables en la prevalencia de estos desórdenes. A mediados de la década de 1980, Flint y colegas observaron frecuencias de α+-talasemia que variaban del 6 al 68% a lo largo de las islas melanesias. Se describieron heterogeneidades similares más tarde en Vanuatu.
En una encuesta de micro-mapeo reciente llevada a cabo en Sri Lanka, la prevalencia de α-talasemia osciló entre el 2 y el 20% (Weatherall DJ, y col.: datos inéditos). Esta variabilidad es probable que sea el resultado de una serie de factores ambientales complejos, incluyendo la tasa de transmisión de la malaria. Una mejor comprensión de estas interacciones ayudará considerablemente en la refinación de las estimaciones de las poblaciones afectadas en las regiones tropicales y en las políticas de desarrollo para el diagnóstico y manejo de la α-talasemia en base a las características de la población local.
Es necesaria una cuantificación precisa de las poblaciones en riesgo de desarrollar síndromes de α-talasemia a nivel nacional, regional, y mundial para definir recursos actuales y futuros requeridos para proporcionar un diagnóstico prenatal preciso de variantes de α-talasemia, un manejo de emergencia de los embarazos con hidropesía fetal por hemoglobina de Bart y sus complicaciones maternas asociadas, y un manejo a largo plazo de la enfermedad por HbH.
Detección
Los objetivos principales de los programas de detección de talasemia son determinar la frecuencia de las diferentes variantes genéticas observadas en las comunidades e identificar e informar a las parejas que están en riesgo, en particular para las formas graves de la enfermedad presentes en las zonas de alta frecuencia.
La detección de la α-talasemia es especialmente útil en la prevención de complicaciones maternas graves en el caso de hidropesía fetal por hemoglobina de Bart, en la provisión de un diagnóstico preciso en los casos en los que la α-talasemia se cohereda con la hemoglobina S o la β-talasemia, o en casos en los que se detecta deficiencia de hierro. La mayoría de los encuestas de detección de α-talasemia son llevadas a cabo como parte de un programa de prevención de β-talasemia y por lo tanto no son adecuadas para determinar frecuencias de α-talasemia en la población.
Los beneficios del cribado de la población deberán considerarse cuidadosamente en cualquier población en la que las variantes de α-talasemia son frecuentes y cuando se observan casos de anemia microcítica hipocrómica inexplicable en ausencia de deficiencia de hierro.
Carga sanitaria y económica
Los primeros estudios sobre la carga global de los trastornos de la hemoglobina que evaluaron la frecuencia de los mismos y el número de años de vida ajustados por discapacidad (AVAD) asociado con ellos se vieron limitados por la falta de datos sobre la α-talasemia, en particular con respecto a la hidropesía fetal de la hemoglobina de Bart y a su carga en relación con los mortinatos o fallecidos poco después del parto.
La Organización Mundial de la Salud (OMS) no recopila datos sobre los mortinatos, y los únicos datos disponibles proveyeron una estimación de 1.250 embarazos con α0-talasemia homocigota por año en Tailandia, una cifra que corresponde a 37.242 AVAD. En comparación, se estimó que la β-talasemia homocigota y la hemoglobina E/β-talasemia resultaron en un total de 53.600 AVAD en Tailandia, en base a una esperanza de vida de 10 años y 30 años, respectivamente. Estas son probablemente subestimaciones porque se consideró la discapacidad de la madre sólo en el último trimestre del embarazo, sin tener en cuenta las complicaciones postnatales que pueden resultar en muerte, y porque no se calcularon las estimaciones para la enfermedad por HbH, ya que se realizó la recolección de los datos antes de que ocurriera una forma particularmente severa de la enfermedad por HbH en muchos países asiáticos.
La información sobre la incidencia de α-talasemia fue insuficiente para permitir el cálculo de la carga regional y global de enfermedad. Se han reportado recientemente estimaciones globales del número de AVAD resultantes de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad del 2010, aunque aún no se han añadido a este proyecto datos precisos sobre los diversos subtipos de talasemias.
Para el conocimiento de los autores, hay un solo estudio hasta la fecha que ha investigado la relación costo-beneficio de un programa de prevención de α-talasemia, establecido por sí mismo o como parte de un amplio programa para todas las talasemias o para condiciones genéticas (por ejemplo, fenilcetonuria) en general. El estudio, llevado a cabo en Hong Kong, concluyó que el cribado prenatal universal para α- y β-talasemias con el uso de pruebas de ADN era rentable, aunque no se dispuso de datos publicados sobre el costo del manejo de un embarazo afectado por hidropesía fetal secundaria a hemoglobina de Bart.
Debe investigarse cuidadosamente si estos resultados son aplicables a países con mayores poblaciones y menores ingresos que los de Hong Kong. Los beneficios del diagnóstico preciso de la deficiencia de hierro y otras hemoglobinopatías - Hemoglobina S y β-talasemia en particular - también deben ser considerados. Los análisis de costo-beneficio de los programas de prevención de β-talasemia en Quebec, Irán, Israel, y el Reino Unido generalmente han confirmado los beneficios generales de este tipo de programas (es decir, los costos de prevención fueron inferiores a los costos de tratamiento), pero los estudios no incluyeron a la α-talasemia.
En los países de bajos y medianos ingresos, esos programas podrían beneficiarse de las infraestructuras existentes (por ejemplo, los programas de cribado de fenilcetonuria o de deficiencia de glucosa-6-fosfato deshidrogenasa en curso) o podrían resultar en una mejora general del acceso a la atención de salud para las comunidades locales.
Manejo
El conocimiento detallado de la prevalencia de la α-talasemia (incluyendo el estado de portador) y de su diversidad genética es esencial para definir políticas orientadas a la reducción de la carga sanitaria a largo plazo de las hemoglobinopatías, permitiendo el diagnóstico preciso (y por lo tanto evitando investigaciones inexactas y caras), el establecimiento de la verdadera causa de la microcitosis, el asesoramiento genético adecuado, y la asignación de recursos para hacer frente a las emergencias y a las necesidades a largo plazo de los pacientes y sus familiares de una manera rentable. Para lograr estos objetivos, la comunidad médica se enfrenta a varios desafíos importantes, que se resumen en la Tabla 1.
Tabla 1. Principales desafíos asociados con la carga sanitaria en aumento de la α-talasemia y recomendaciones para superarlos
En ausencia de tratamientos específicos y de una clara comprensión de los mecanismos subyacentes responsables de la amplia gama de anomalías congénitas asociadas con este trastorno letal, el cribado y el diagnóstico prenatal temprano representan las únicas opciones para identificar los embarazos de alto riesgo y evitar complicaciones maternas graves.
Aunque por lo general se recomienda la interrupción de los embarazos afectados debido al aumento del riesgo de complicaciones maternas y fetales severas y a los efectos psicológicos en las familias, deben tenerse en cuenta los antecedentes culturales y religiosos cuando se asesora a las parejas en riesgo, tanto en las comunidades en las que la α-talasemia ha sido prevalente tradicionalmente como en aquellas en las que se ha introducido recientemente a través de la migración.
Un diagnóstico temprano correcto en el embarazo es esencial para evitar complicaciones médicas graves y trauma psicológico. Tal diagnóstico puede hacerse de forma confiable con costos relativamente bajos previendo que las mujeres embarazadas reciban un seguimiento regular por personal médico familiarizado con este síndrome.
Los recientes progresos en las pruebas de ADN revelaron una mayor diversidad fenotípica de la enfermedad por HbH que lo que se pensaba previamente.
Como se ha señalado anteriormente, las variantes no delecionales suelen ser más graves que las variantes de deleción. Por ello es crucial realizar el análisis del ADN para identificar la variante genética subyacente responsable de este trastorno. La reacción en cadena de la polimerasa-Gap (PCR) y los ensayos de PCR multiplex permiten una fácil detección de una gama de variantes comunes, pero tales métodos siguen siendo costosos y no son ampliamente utilizados en países de bajos y medianos ingresos. Con los métodos de prueba de ADN cada vez más y más asequibles, es probable que éstos se conviertan en una parte regular de los servicios disponibles para el control y manejo de las talasemias, en particular en los países asiáticos.
A medida que el conocimiento sobre la relación fenotipo-genotipo en la enfermedad por HbH mejora, la correcta identificación de genotipos será imprescindible para informar a los padres sobre los riesgos reproductivos durante el asesoramiento genético y para dar un adecuado cuidado a los pacientes afectados.
Con el aumento de los movimientos de población, la diversidad de combinaciones de variantes de α-talasemia o hemoglobinopatías coheredadas seguirá aumentando. Aunque es difícil predecir el fenotipo exacto de estas nuevas combinaciones, un conocimiento detallado de la distribución actual de las variantes genéticas ayudará al menos en la definición de los procedimientos de diagnóstico y en la evaluación de los riesgos potenciales.
Las colaboraciones entre las Áreas de "Origen" (aquellas con una alta prevalencia de α-talasemia y emigración sustancial) y las Áreas "Sumidero" (las que tienen inmigración sustancial de las zonas de origen), como las desarrolladas entre Filipinas y California y entre Sri Lanka y el Reino Unido, sin duda representan modelos beneficiosos para ambas partes y deben ser replicados más ampliamente.
La carga sanitaria de la α+-talasemia actualmente no se conoce. Aunque las variantes de α+-talasemia por si solas no representan un problema clínico directo, son probablemente los trastornos genéticos más comunes en el mundo, y representan importantes modificadores genéticos para una serie de condiciones, incluyendo la malaria, los trastornos de células falciformes, la β-talasemia, y la deficiencia de hierro.
Una mejor comprensión de estas interacciones es crucial para proporcionar un diagnóstico y tratamiento adecuado para los pacientes afectados, pero será relevante sólo si las intervenciones resultantes de esta mejor comprensión se implementan en todas las áreas en las que estas interacciones se están produciendo. Por ejemplo, se desconoce actualmente si las interacciones epistáticas entre el rasgo de células falciformes y la α+-talasemia observadas en Kenia también se están produciendo en las poblaciones indígenas.
Debido a la notable heterogeneidad geográfica en la prevalencia de la α-talasemia, las intervenciones tienen que adaptarse a las características específicas de la población local (por ejemplo, prevalencia del trastorno en la población, composición étnica, y consanguinidad) y al sistema de salud local. Una mejor comprensión de la relación entre la prevalencia de las variantes de α-talasemia, los factores ambientales y las infecciones, en combinación con métodos analíticos y de modelización modernos, ayudaría a lograr estimaciones más refinadas de las poblaciones afectadas. Tal conocimiento también ayudaría en el desarrollo de programas de prevención estratificados enfocados, por ejemplo, en regiones en las que las variantes de α0-talasemia son más frecuentes.
Conclusiones
Las α-talasemias representan un problema de salud mundial con una carga creciente. Un conocimiento refinado de las bases moleculares de la α-talasemia será totalmente relevante desde la perspectiva de la salud pública sólo si se complementa con datos epidemiológicos detallados.
Para garantizar el adecuado cuidado de los pacientes y la sostenibilidad de los sistemas de atención de salud, se debe poner mayor esfuerzo en obtener estimaciones basadas en la evidencia de las poblaciones afectadas, la provisión de recursos para la prevención, control y manejo de las talasemias, y realizar análisis de costo-efectividad. Tales objetivos se alcanzarán sólo a través de un esfuerzo concertado de las comunidades de investigación y médicas y el apoyo de agencias de financiación internacional para recopilar y compartir datos epidemiológicos. La reciente inclusión de las hemoglobinopatías en el Estudio de Carga Mundial de Enfermedad y la evaluación de la carga de la α-talasemia en términos de AVAD tendrán sentido sólo si se dispone de datos confiables y actualizados.
Comentario:
Las talasemias son trastornos hereditarios de la síntesis de la hemoglobina que se caracterizan por una producción reducida de las cadenas de globina. Presentan una amplia distribución mundial debido al movimiento poblacional constante, pero no se cuenta actualmente con datos exactos de prevalencia. Estas migraciones de población contribuyen a la propagación de estas hemoglobinopatías aumentando el número de países que requieren la aplicación de intervenciones específicas para educar a sus comunidades, diagnosticar los trastornos, y asesorar a los pacientes afectados y a sus familias. Se requieren esfuerzos conjuntos para determinar las bases moleculares de las variantes de talasemia y su epidemiología a nivel global y regional, y para desarrollar programas de detección, manejo y asesoramiento con respecto a estos trastornos particulares de la hemoglobina.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol
jueves, 19 de marzo de 2015
Viletanos. El Triple Whammy.
Gemma Arrufat (R3 Farmacia Hospitalaria), nos habló de el Triple Whammy.
Se trata del riesgo renal asociado a la administración concomitante de
tres clases de fármacos: diuréticos, IECAs/ARA-II y AINEs.
Puntos Clave:
Enlaces de Interés:
Puntos Clave:
- Siempre que sea posible, evitar AINEs en pacientes con IRC, ICC y HTA, sobretodo si estos pacientes están tomando IECAs y/o diuréticos.
- La combinación de la triple terapia se asoció a un riesgo de fallo renal agudo (RR 1,31, IC 95%(1,12-1,53)).
- Los fármacos antihipertensivos tienen sus beneficios cardiovasculares, pero se debe vigilar cuando se utilizan concomitantemente con AINEs.
Enlaces de Interés:
- Dosificación
de Medicamentos en la Enfermedad Renal Crónica. Infac. Volumen
22-Nº1/2. 2014. [Citado el 18-3-2015]. Disponible en internet en: http://www.osakidetza.
euskadi.eus/contenidos/ informacion/cevime_infac_2014/ es_def/adjuntos/INFAC_Vol_22_ 1_2_Enfermedad_renal_cronica. pdf
martes, 17 de marzo de 2015
Cosas del PAC. Un repaso al edema agudo de pulmón
Atendimos hace unos días a un paciente varón de setenta y tantos años al
que la ambulancia sanitarizada trasladaba desde una población cercana
al hospital. Se trataba de un paciente con HTA, EPOC y portador de
marcapasos (entre otras...) que había consultado por disnea de unos días
de evolución. Durante el traslado en ambulancia su disnea empeoró
muchísimo y siguiendo la indicación del médico coordinador pararon en
nuestro PAC. También, por indicación desde coordinación, le habían
administrado una nebulización de salbutamol sin que experimentara
mejoría.
El hombre estaba muy afectado, taquipnéico, con importante trabajo
respiratorio y mal perfundido, no podía ni hablar. El saturómetro
señalaba una cifra que era mejor ignorar, para no agobiarnos más;
mantenía cifras de PA altas, estaba taquicárdico, en la auscultación
pulmonar se escuchaban roncus y crepitantes en ambos campos sin
sibilantes (tampoco los había antes de la nebulización, a juicio del
enfermero que le había auscultado) y tenía unos mínimos edemas
pretibiales. No parecía que hubiera historia de dolor torácico, aunque
sí nos explicó entrecortadamente que notaba una sensación de tope en
epigastrio, ni clínica clara de infección respiratoria días previos ni
marcada disminución de diuresis. El ECG ayudaba poco: ritmo de
marcapasos y abundantes extras ventriculares multifocales...
Disnea: ¿cardíaca? ¿respiratoria? ¿ambas cosas a la vez? Nos parecía más de origen cardíaco, pero...
Con la sospecha de insuficiencia cardíaca aguda/edema de pulmón, optamos por:
- Sedestación, ECG, monitorización, toma de constantes y exploración
- Oxígeno: a pesar de su EPOC de entrada 100%, cuando conseguimos saturaciones seguras (90-92%), bajamos la concentración
- Vía con fisio (solo mantener vía)
- Furosemida: 40 mg iv
- Solinitrina: 2 pufs
- Cloruro mórfico: 1amp al 1%+9 de fisio, pasados 2 cc iv
- Y además, para completar el kit respiratorio que no lo teníamos claro: 40 mg de metilprednisolona iv
Con todo, nuestro paciente mejoró mucho y procedimos a su traslado en medicalizada.
Ya con tranquilidad, en casa, me puse a trastear y a repasar el manejo
del Edema Agudo de Pulmón (EAP), porque aunque no tuve muchas dudas
quería comprobar que no me hubiera dejado nada importante.
Y claro: sorpresas...
Os pongo un cuadro con los aspectos que, en nuestro ámbito, son más controvertidos.
Os pongo un cuadro con los aspectos que, en nuestro ámbito, son más controvertidos.
Fuente de información
|
Morfina
|
Nitratos
|
| UpToDate (enero 2015) Treatment of acute decompensated heart failure: Components of therapy |
No administrar, salvo en contexto de IAM
|
Vía endovenosa, asociados a diuréticos si respuesta
insuficiente y ausencia de hipotensión
|
DynaMed
Acute heart failure |
Considerar en pacientes con disnea severa para disminuir
la ansiedad
|
Vía endovenosa, asociados a diuréticos si respuesta
insuficiente y ausencia de hipotensión
|
No lo contempla en sus directrices
|
Vía endovenosa, asociados a diuréticos si respuesta
insuficiente y ausencia de hipotensión
|
|
Considerar en pacientes con disnea severa para disminuir
la ansiedad (No señalan grado de recomendación)
|
Vía endovenosa, asociados a diuréticos si respuesta
insuficiente y ausencia de hipotensión
|
|
No administrar de rutina
|
No administrar de rutina.
Matizan: puede tener su lugar en IAM concomitante, HTA
severa, regurgitación severa de válvula aórtica o afectación valvular mitral.
Advierten del riesgo de generar hipotensión y aconsejan vía ev con
monitorización constante. Señalan que las pruebas sobre su beneficio son
limitadas.
|
|
Evidencia insuficiente para los resultados clínicos
obtenidos con el uso de nitratos en el fallo cardíaco agudo
|
Algunas matizaciones:
- En relación al uso de opioides: la mayoría señalan que: el sulfato de morfina se utiliza clásicamente para disminuir la ansiedad y el distress respiratorio en estos pacientes y disminuir la precarga. Sin embargo, a la luz de estudios retrospectivos su uso parece favorecer la necesidad de soporte ventilatorio invasivo, se asocia con un aumento de los ingresos en UCI, estancias hospitalarias más prolongadas y mayor mortalidad aunque esta última afirmación no puede asegurarse como una causa/efecto de forma contundente. Advierten además de los posibles efectos adversos como la aparición de náuseas o la depresión del centro respiratorio. Reconocen la falta de estudios de calidad que analicen el papel de los opioides en el EAP, así como la dificultad de realizar ensayos clínicos en este contexto por razones éticas. Un estudio sugiere el uso de benzodiacepinas en lugar de la morfina para el tratamiento de la ansiedad en estos casos, argumentando que son más eficaces y producen menos depresión respiratoria.
- En relación al uso de nitratos: en sus recomendaciones lo señalan como un tratamiento coadyuvante añadido al uso de diuréticos iv para aquellos casos en que sean necesarios y en pacientes con cifras tensionales altas. En cuanto a la vía de administración, contemplan la endovenosa con monitorización que permita un adecuado ajuste de dosis. Únicamente en la guía de NICE entre los estudios que analizan incluyen un ensayo clínico que etiquetan de muy baja calidad, con un reducido número de pacientes (20) y en el contexto de fallo agudo asociado a IAM y en el que comparan la administración de dinitrato de isosorbide oral frente a placebo; las variables que miden son hemodinámicas y no está claro si los beneficios en estos parámetros se traducen en una mejoría clínica.
- Finalmente, por aquello de que es una práctica bastante extendida (¿o no?) la de hacer tercio y mitad cuando tenemos duda de si la disnea es cardíaca, respiratoria o mixta, en DynaMed señalan que el uso de broncodilatadores nebulizados en pacientes con fallo cardíaco agudo y sin historia de EPOC se asocia con una mayor necesidad de ventilación mecánica y de vasodilatadores endovenosos.
Hasta
aquí llego. Ahora queda formularse uno mismo las preguntas. A ciencia
cierta, no sé qué haré ante una situación de este tipo en próximas
ocasiones; puede que haga exactamente lo mismo, puede que deje de hacer
algunas cosas, puede que piense antes para qué las hago (¿tal vez alguna
de ellas las haga más para mí que para nadie...?). En todo caso, doy
por bien empleado el tiempo invertido en este post si os sirve para
repasar el tema y reflexionar...Nadie dijo que esto fuera fácil...
Ágora Docente. HOMBRO DOLOROSO. Aproximación al manejo en atención primaria.
Hombro doloroso from Unitat Docent de Medicina Familiar i Comunitària de Menorca
citas bibliográficas:
citas bibliográficas:
| Dolor de hombro en la consulta de atención primaria. C. Alba romero. FMC. 2014;21(7):404-10 |
| Miembro superior: hombro: síndrome subacromial. AMF 2008;4(5):244-252 |
| Hombro doloroso. A tejedor. AMF 2005;1(2):63-74 |
| Evaluation of the patient with shoulder complaints. Bruce C Anderson, MD. UpToDate. Topic 238 v 23 Feb 2015 |
| Chronic shoulder pain. Kelton M et all. Am Fam Physician, 2008 Fef 15;77(4):453-460 ; 493-497 |
| Páginas web :www.fisioterapiatualcance.es www.sermef-ejercicios.org |
| Curso de fisioterapia y rehabilitación. FMC Curso 2009;16 (Supl. 1):1 |
| Procedimientos terapéuticos en fisioterapia y traumatología en atención primaria (II). FMC 2005 |
Sapiens Medicus. Cómo interpretar una gasometría en 5 pasos.
 La gasometría es un estudio invasivo en el que se punciona una
arteria para obtener una muestra de sangre arterial. Después de ser
procesada en el laboratorio es utilizada para determinar los valores
sanguíneos de pH, PO2, PCO2 y HCO3, entre otras cosas. Esto la
convierte en una herramienta indispensable para el diagnóstico y
seguimiento de patologías que trastornen el equilibrio ácido-base en los
líquidos corporales, las cuales son el pan de cada día en los servicios
de medicina interna y urgencias, y tú… ¿sabes cómo interpretarla?
La gasometría es un estudio invasivo en el que se punciona una
arteria para obtener una muestra de sangre arterial. Después de ser
procesada en el laboratorio es utilizada para determinar los valores
sanguíneos de pH, PO2, PCO2 y HCO3, entre otras cosas. Esto la
convierte en una herramienta indispensable para el diagnóstico y
seguimiento de patologías que trastornen el equilibrio ácido-base en los
líquidos corporales, las cuales son el pan de cada día en los servicios
de medicina interna y urgencias, y tú… ¿sabes cómo interpretarla?Conceptos que debes saber
El potencial de hidrógeno, o pH se refiere al potencial de una molécula para liberar o absorber iones hidrógeno (H+) (o hidrogeniones) al estar en una solución. Se determina utilizando un pHmetro o la siguiente fórmula: pH= -log 10 (aH+), podría ahondar más en ello pero no es el tema, así que prosigamos. El desequilibrio ácido-base es la alteración de la relación fisiológica entre los hidrogeniones y sus “buffers” o amortiguadores en la sangre, que tiene repercusión clínica en el paciente y laboratorial en el pH sanguíneo.Cuando hablamos de buffers (o amortiguadores) nos referimos a moléculas que el cuerpo produce para mantener balanceado el pH sanguíneo. Existen múltiples “buffers” como la hemoglobina, el fosfato, etc., pero el ejemplo clásico y más conocido, es el buffer de bicarbonato-ácido carbónico, que funciona captando hidrogeniones cuando es bicarbonato y liberándolos cuando es ácido carbónico.
Parámetros normales y terminología
- pH: 7.35-7.45
- PCO2: 35-45 mmHg
- Cationes:
- Na+ 135-145 mEq/l
- K+ 3.5-4.5 mEq/l
- Aniones:
- HCO3 24-28 mEq/l
- Cl- 99-104 mEq/l
- Brecha aniónica (anion gap, AG) 3-10 [AG=Na-(Cl+HCO3)]
- Albúmina: 4 g/dl
- Acidemia: pH <7.35
- Alcalemia: pH>7.45
- Acidosis: H+ séricos elevados
- Alcalosis: H+ séricos disminuidos
- A… Respiratoria: alt. en la PCO2
- A… Metabólica: alt. en el HCO3
Sigue leyendo......http://sapiensmedicus.org/blog/2014/09/28/como-interpretar-una-gasometria-en-5-pasos/
jueves, 12 de marzo de 2015
Guía NICE. Neumonia adquirida en la comunidad y hospitalaria. Fuente IntraMed.
Autor: Sinan Eccles, Celia Pincus, Bernard Higgins, Mark Woodhead BMJ 2014;349:g6722. Diagnosis and management of community and hospital acquired pneumonia in adults: summary of NICE guidance
La neumonía adquirida en la comunidad es
una enfermedad que causa una elevada morbilidad y tiene una
probabilidad de mortalidad del 20 % en los pacientes admitidos en
hospitales de Inglaterra.Se diagnostica en 5 a 12% de los adultos con síntomas de infección de la vía área respiratoria inferior y de ellos un 22 a 42% son internados.
La neumonía adquirida en el hospital (excluyendo la neumonía asociada al ventilador) tiene una prevalencia de aproximadamente un 1% entre los pacientes internados. Se estima que puede demorar el ingreso al hospital unos 8 días y tiene una elevada tasa de mortalidad.
Este artículo resume las más recientes recomendaciones de las guías NICE para el manejo de ambos tipos de neumonía.
Recomendaciones:
Estas recomendaciones se basan en revisiones sistemáticas de la mejor evidencia disponible y ponen especial consideración en el costo – beneficio. Cuando la evidencia disponible es mínima, la recomendación se basan en la experiencia y opinión de la buena práctica del Guideline Development Group.
Presentación con infección respiratoria baja:
De este grupo de pacientes solo una menor cantidad tienen neumonía. En aquellos que no tienen el diagnóstico clínico de neumonía, la decisión de prescribir antibióticos puede ser difícil con tendencia a la sobremedicación. La Proteína C Reactiva (PCR) puede ayudar a identificar pacientes con infección respiratoria de vías inferiores que se beneficiaran o no con antibióticos.
• En atención primaria, en personas con síntomas de infección respiratoria baja considere realizar PCR si luego de la evaluación clínica no se ha llegado al diagnóstico de neumonía y si no está claro si deben ser indicados antibióticos. Utilice los resultados del test para guiarse en la prescripción antibiótica en personas sin diagnóstico clínico de neumonía de la siguiente manera:
1. No indique antibióticos de rutina si la concentración de PCR es menor a 20mg/L.
2. Considere demorar la toma de antibióticos (utilizarlos solo si los síntomas empeoran mas tarde) si la concentración de PCR se encuentra entre 20 y 100mg/L.
3. Indique antibióticos si la concentración de PCR es mayor a 100mg/L.
(Basado en alta a muy baja calidad de evidencia a partir de ensayos controlados randomizados con un gran número de pacientes un análisis costo efectivo)
Neumonía adquirida en la comunidad:
Es muy importante evaluar la severidad de la enfermedad para guiar luego el tratamiento antibiótico y la posibilidad de internación.
Evaluación de la severidad en atención primaria.
• Luego del diagnóstico, evalúe si el paciente se encuentra en un riesgo de muerte bajo, intermedio o moderado, utilizando el score CRB65 (ver tabla 1).
• Utilice el juicio clínico junto al score CRB65 para decidir si el paciente necesita permanecer en el hospital, como se muestra a continuación:
→ Con un CRB65 de 0, considere cuidado en el hogar.
→ Para el resto de los pacientes considere la internación, especialmente aquellos con un score CRB65 mayor a 2.
(Basado en la información observacional limitada y la experiencia y opinión de GDG)
Tabla 1: Score para la evaluación del riesgo en atención primaria
|
El score CRB65 se calcula asignando 1 punto a cada una de las siguientes caracteristicas: • Confusión • Frecuencia respiratoria aumentada (mayor a 30 ventilaciones por minuto) • Presión arterial baja (PA diastólica de 60 mmHg o menor y PA sistólica menor a 90 mmHg) • Edad mayor a 65 años
Los pacientes se estratifican por riesgo de muerte de la siguiente manera:
• 0 = bajo riesgo (riesgo de mortalidad menor al 1%) • 1 o 2= riesgo intermedio (riesgo de mortalidad del 1 al 10%) • 3 o 4= riesgo elevado (riesgo de mortalidad mayor al 10%) |
Cuando el diagnóstico de neumonía adquirida de la comunidad se hace en el hospital, evalúe si el paciente se encuentra en un riesgo de muerte bajo, intermedio o moderado, utilizando el score CURB65, ver tabla 2.
• Utilice el juicio clínico junto al score CURB65 para evaluar el manejo, como se muestra a continuación:
→ En pacientes con un score CURB65 de 0 a 1, considere cuidados en el hogar.
→ En pacientes con un score CURB65 de 2 o mayor, considere hospitalización.
→ En pacientes con un score CURB65 de 3 o más considere internación en UTI.
(Basado en estudios de cohorte de moderada a muy baja calidad con un gran número de pacientes y la experiencia y opinión de GDG)
Estratifique a los pacientes con neumonía adquirida de la comunidad en baja, moderada o alta severidad. El grado de severidad usualmente se corresponderá con el riesgo de muerte. (Basado en la experiencia y opinión del GDG).
Tabla 2: Score CURB65 para la evaluación del riesgo de mortalidad en el hospital:
|
El score CRB65 se calcula asignando 1 punto a cada una de las siguientes caracteristicas: • Confusión • Aumento de la uremia (mayor a 7mmol/L) • Frecuencia respiratorio aumentada (mayor a 30 ventilaciones por minuto) • Presion arterial baja (PA diastolica de 60 mmHg o menor y PA sistolica menor a 90 mmHg) • Edad mayor a 65 años Los pacientes se estratifican por riesgo de muerte de la siguiente manera: • 0 a 1 = bajor riesgo (riesgo de mortalidad menor al 3%) • 2 = riesgo intermedio (riesgo de mortalidad del 3 al 15%) • 3 a 5 = riesgo elevado ( riesgo de mortalidad mayor al 15%) |
Test microbiológicos:
No ofrezca en forma rutinaria test microbiológicos a los pacientes con neumonía adquirida en la comunidad de severidad baja.
En aquellos con enfermedad de severidad moderada a alta debe:
Tomar cultivos de sangre y esputos y considere los test de antígeno urinario para neumococo y legionella.
(Basado en evidencia de baja y muy baja calidad de evidencia a partir de estudios randomizados y no randomizados, un estudio económico original y la experiencia y opinión del GDG.)
Diagnóstico y tratamiento oportuno:
La administración temprana de antibióticos en pacientes internados con neumonía adquirida de la comunidad mejora los resultados pero debe asociarse a un rápido y adecuado diagnóstico para evitar una administración inapropiada y dañina de antibióticos a aquellos que en realidad tienen otro diagnóstico (por ejemplo: insuficiencia cardíaca).
Realice estudios diagnósticos (incluídos Rayos X) para llegar al diagnóstico y tratamiento dentro de las 4 horas de ingreso al hospital.
Luego del diagnóstico comience el tratamiento antibiótico lo antes posible dentro de las 4 primeras horas de ingreso.
(Basado en estudios de cohorte observacional de baja y muy baja calidad con un gran número de pacientes)
Antibióticoterapia:
Los antibióticos son la piedra fundamental del tratamiento de la neumonía, pero pueden causar daño.
Actualmente se recomienda una duración de 5 días de tratamiento en neumonias de severidad baja, esta es menor que en previas recomendaciones.
Esta recomendación incluye una advertencia de seguridad para los pacientes a los que sugiere buscar consejo médico si no sienten mejoras y a los médicos clínicos les sugiere extender el tratamiento si los resultados no son los esperados.
El uso de antibióticos por tiempo prolongado, así como la terapia con dos fármacos deber reservarse para pacientes con neumonía adquirida de la comunidad de moderada a alta severidad.
Neumonía adquirida en la comunidad de baja severidad:
• Indique 5 días de antibióticos en estos pacientes.
(Basado en moderada a muy baja calidad de evidencia obtenida a partir de ensayos controlados randomizados, un análisis de costos con limitaciones y la opinión y experiencia del GDG)
• Prefiera amoxicilina en lugar de macrólido o tetraciclina. Considere estos últimos en pacientes alérgicos a la penicilina.
(Basado en evidencia inconclusa obtenida a partir de estudios controlados randomizados y la experiencia y opinión del GDG)
• Si luego de tres días de tratamiento el paciente no evoluciona como es esperado, considere extender el tratamiento a más de 5 días. (Basado en la experiencia y opinión del GDG).
• Debe explicarle al paciente y/o sus familiares y cuidadores que es necesario que busquen atención médica si luego de tres días de tratamiento no se registran mejoras o si los síntomas empeoran antes de este periodo. (Basado en la experiencia y opinión del GDG).
• No utilice de rutina:
- Una fluoroquinolona
- Terapia antibiótica dual
Neumonía adquirida en la comunidad de severidad moderada a alta:
• Considere una duración de 7 a 10 días de la terapia antibiótica en estos pacientes.
(Basado en la experiencia y opinión del GDG)
• En la enfermedad de severidad moderada considere la utilización de terapia dual con amoxicilina y un macrólido.
• En pacientes con neumonía de severidad alta considere la terapia antibiótica dual con beta lactámicos y macrólidos, como por ejemplo: amoxicilina clavulánico, cefotaxima, ceftarolina fosamil, ceftriaxona, cefurexima y piperacilina/tazobactam.
(Basado en moderada a muy baja calidad de ensayos controlados randomizados y baja a muy baja calidad de evidencia a partir de estudios de cohorte observacional, con una gran cantidad de pacientes, análisis de costo efectividad y la experiencia y opinión de GDG)
Tratamiento con glucocorticoides:
No administrar de rutina salvo en casos que el paciente presente una condición de base que los requiera.
(Basado en moderada a muy baja calidad de evidencia a partir de ensayos controlados randomizados y la experiencia y opinión de GDG)
Monitoreo en el hospital
En estos pacientes es de utilidad medir la PCR para identificar a aquellos que no responden al tratamiento o necesitan una revisión de su manejo.
• Considere medir la PCR al ingreso y repita el test si no hay un progreso certero dentro de las 48/72hs.
(Basado en baja a muy baja calidad de estudios de cohorte observacionales y la experiencia y opinión del GDG)
Externación segura:
Reducir el tiempo de hospitalización es un objetivo común en el Reino Unido. De todas formas un alta de la internación temprana puede resultar en un aumento de la mortalidad y mayores tasas de readmisión.
• No de el alta a un paciente con neumonía adquirida en la comunidad si en las 24 hs. previas ha tenido 2 o más de los siguientes signos/síntomas:

Muchos pacientes no son informados sobre qué deben esperar una vez recuperados. Conocer el tiempo normal de recuperación podría colaborar a disminuir su ansiedad, así como también advertir sobre la necesidad de buscar consejo médico si no mejoran como es esperable.
• Explique a los pacientes que una vez iniciado el tratamiento los síntomas deben mejorar. La tasa de mejoría varía con el tipo de severidad de la neumonía, pero la mayoría de los pacientes pueden esperar lo siguiente:
1 semana: debería desaparecer la fiebre.
4 semanas: debe reducirse claramente el dolor de pecho y la producción de esputo.
6 semanas: la tos y la falta de aliento deberían reducirse notablemente.
3 meses: la mayoría de los síntomas tendrían que estar resueltos, pero la fatiga podría estar presente.
6 meses: la mayoría de las personas no presentan síntomas asociados al cuadro.
- Aconseje a sus pacientes que consulten a un médico si sus síntomas empeoran o no mejoran como se espera.
Neumonía adquirida en el hospital (excluyendo neumonía asociada al ventilador)
Desafortunadamente la evidencia para esta patología es escasa. Debido a esto el GDG no ha podido realizar recomendaciones específicas en la mayoría de los tópicos examinados.
Terapia antibiótica:
• Instale tratamiento antibiótico lo antes posible luego del diagnóstico, dentro de las 4hs. en pacientes con neumonía hospitalaria. (Basado en la experiencia y opinión del GDG)
• Seleccione el tratamiento antibiótico de acuerdo con la política hospitalaria local (lo que incluye el conocimiento de los microorganismos patógenos locales) y las circunstancias clínicas. (Basado en evidencia de baja a muy baja calidad a partir de ensayos controlados randomizados y la experiencia y opinión del GDG)
• Considere una duración de 5 a 10 días de tratamiento antibiótico. (Basado en la experiencia y opinión del GDG).
Superando barreras:
Muchos pacientes esperan recibir antibióticos cada vez que sienten enfermos con tos productiva, y dos aspectos de estas nuevas recomendaciones serían contrarias a estas expectativas.
Ambos son importantes porque el uso excesivo de antibióticos puede ser perjudicial para algunos pacientes (debido a los efectos adversos de los fármacos y complicaciones como la infección por Clostridium difficile) y para la población en general (debido a la resistencia antibiótica).
La utilización de PCR en atención primaria en una nueva recomendación que va a requerir un costo y educación a lo largo del tiempo.
Sin embargo, incorporar el resultado de la prueba en las discusiones con los pacientes debe ayudar a tranquilizarlos cuando los antibióticos no son indicados (la mayoría de los casos).
Muchas personas que han recibido antibióticos van a recibir un segundo curso porque sus síntomas no han desaparecido por completo. La evidencia sobre la resolución natural esperada de los síntomas sugiere que la mayoría de estos cursos son probablemente innecesarios, y la educación en este punto también debería reducir el uso de antibióticos fuera de lugar.
Suscribirse a:
Comentarios (Atom)